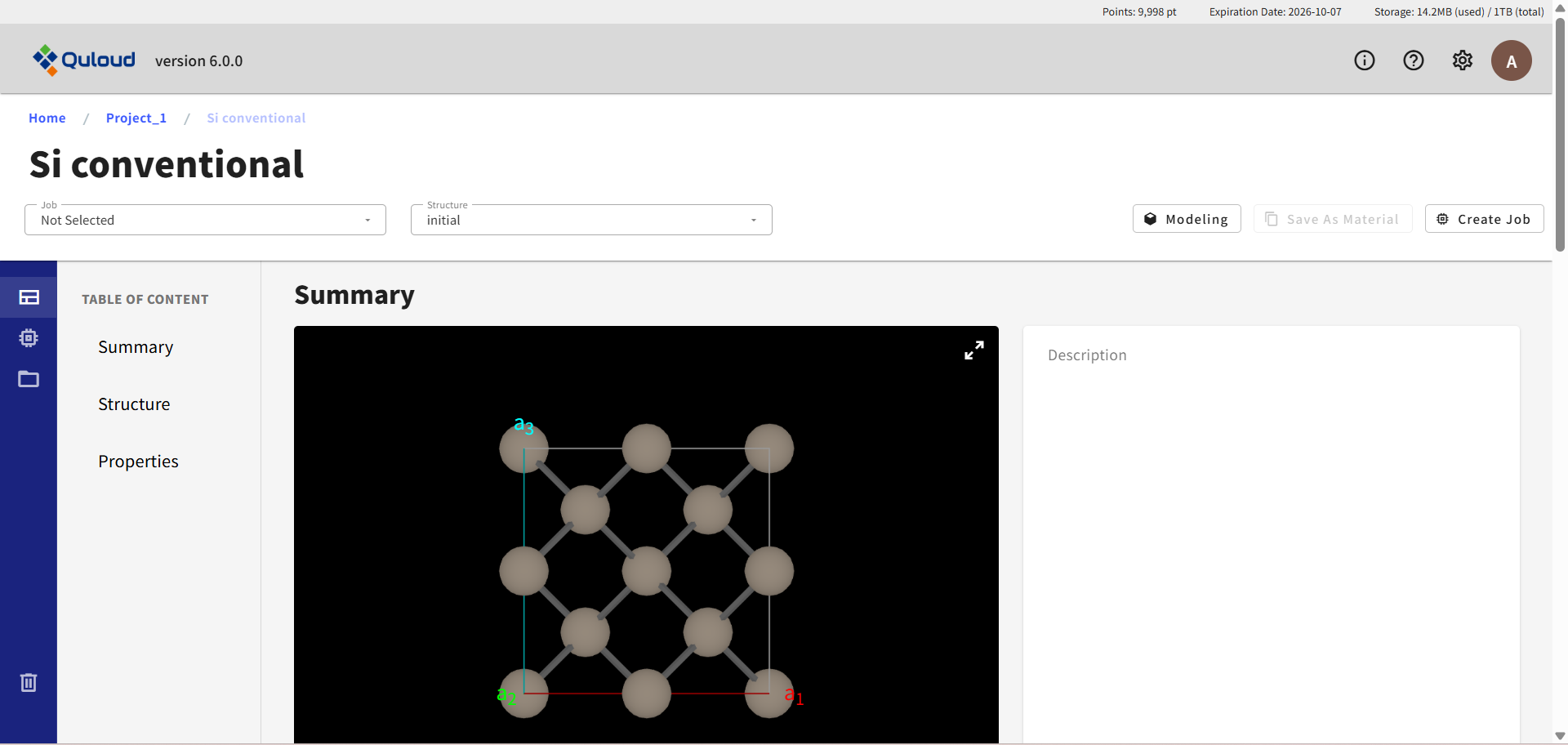
7. Material のモデリング
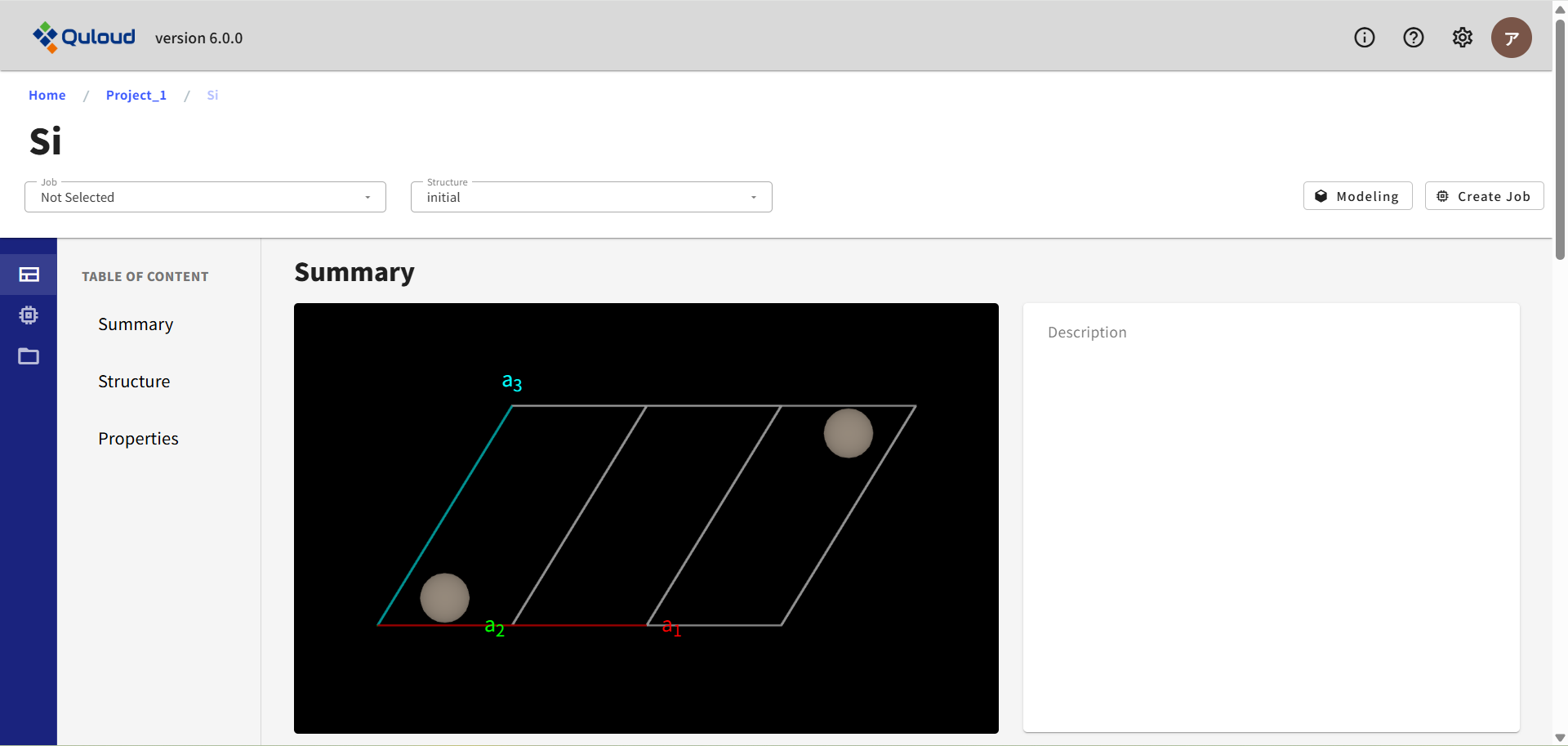
上図のMaterial 詳細画面の上部にある「Modeling」ボタンをクリックすると、 下図のようなモデリング専用の画面がブラウザの別タブとして開きます。 上図では initial(登録された Material)が選択されていますが、 Select Property で、例えば原子構造最適化ジョブの結果などが選ばれていれば、その Property の構造を元にモデリングが開始されます。
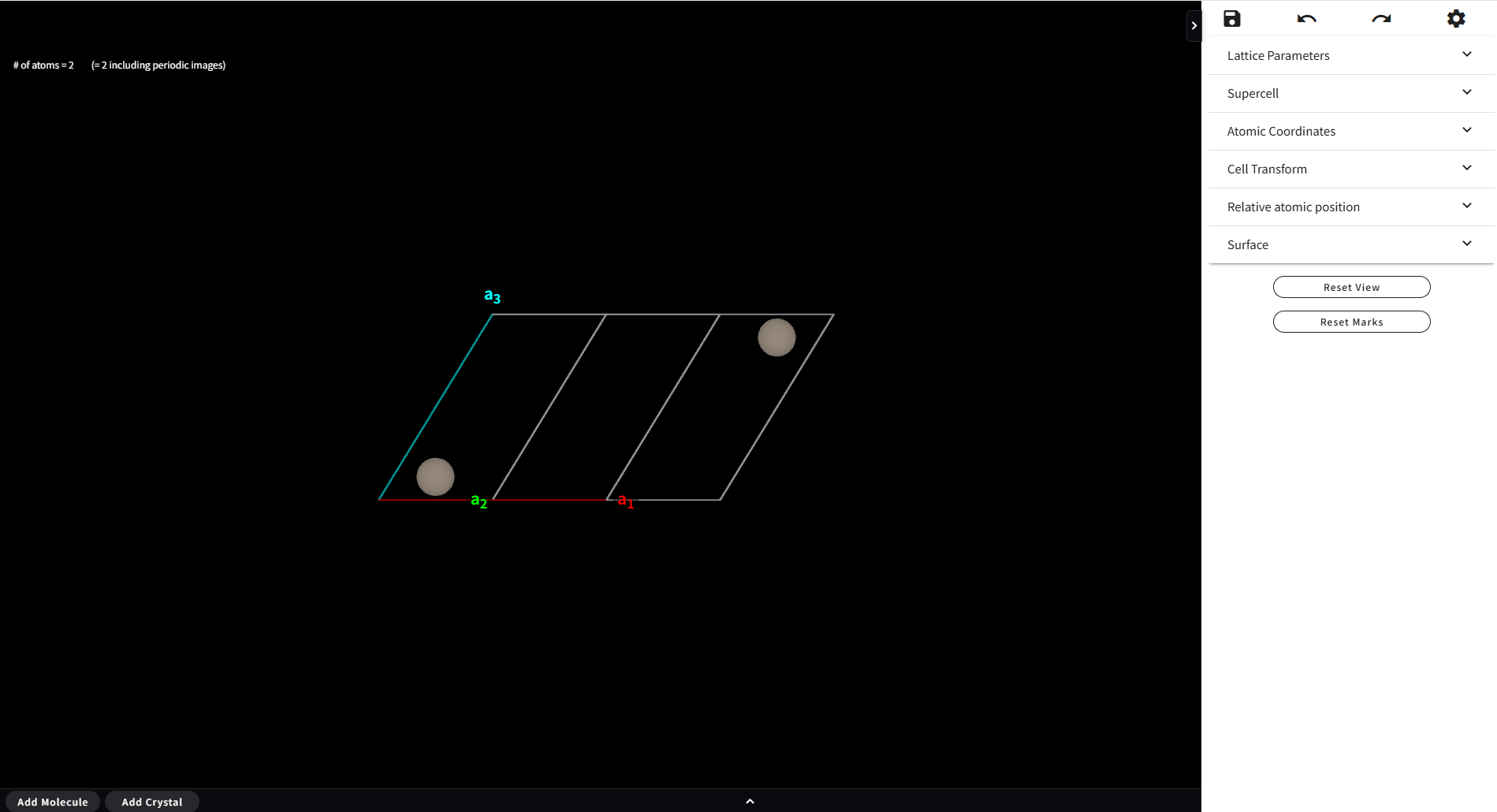
7.1. 基本的な操作
モデリング画面では、以下のマウス操作で原子モデルの回転および拡大・縮小ができます。
回転: ドラッグ
拡大・縮小: マウスホイール
原子がいない場所にマウスカーソルを持ってきて右クリックすると、原子を追加する「Add atom」メニューが現れます。
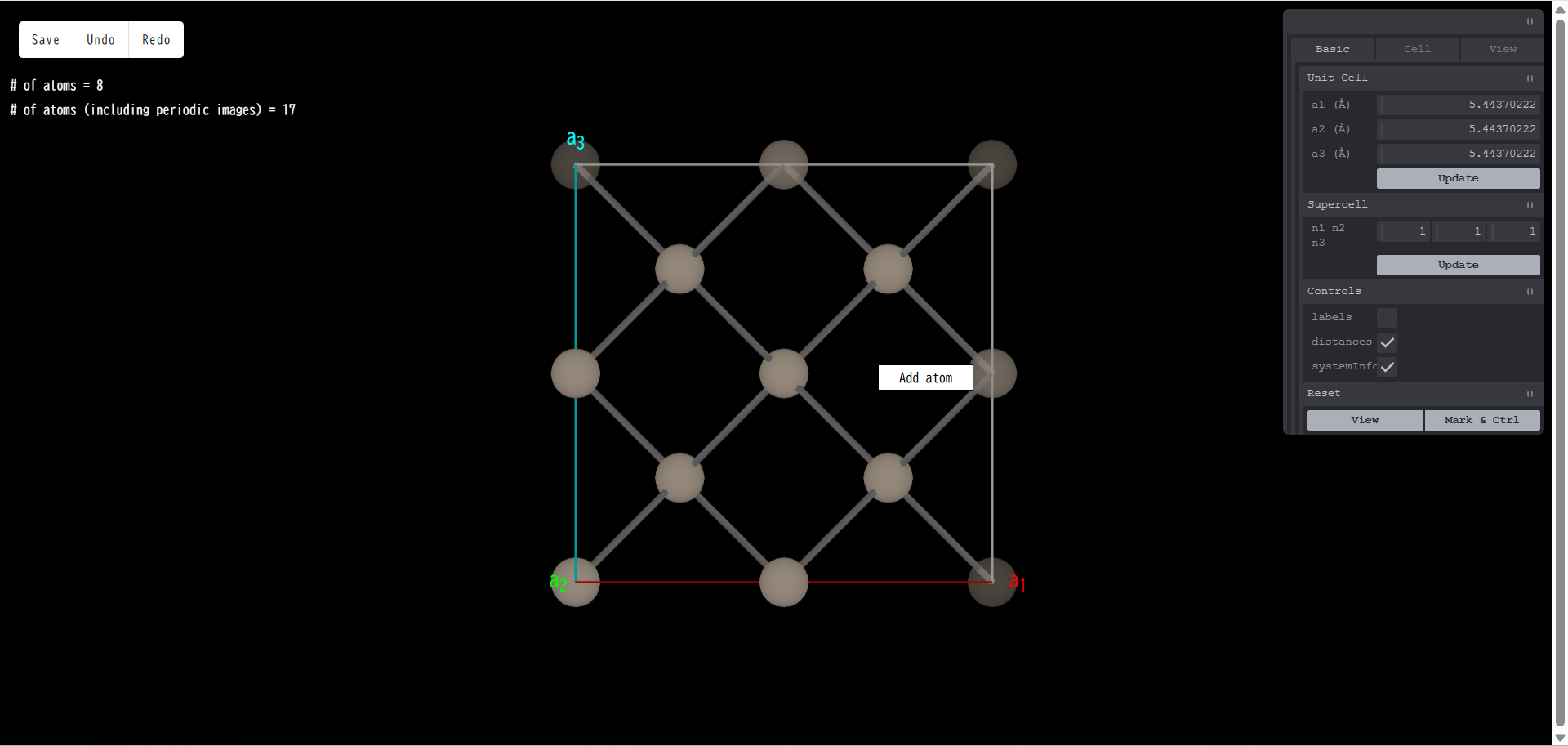
「Add atom」をクリックすると、新しいウィンドウで元素周期表が現れます。ここで、どの元素の原子を追加するか選択します。
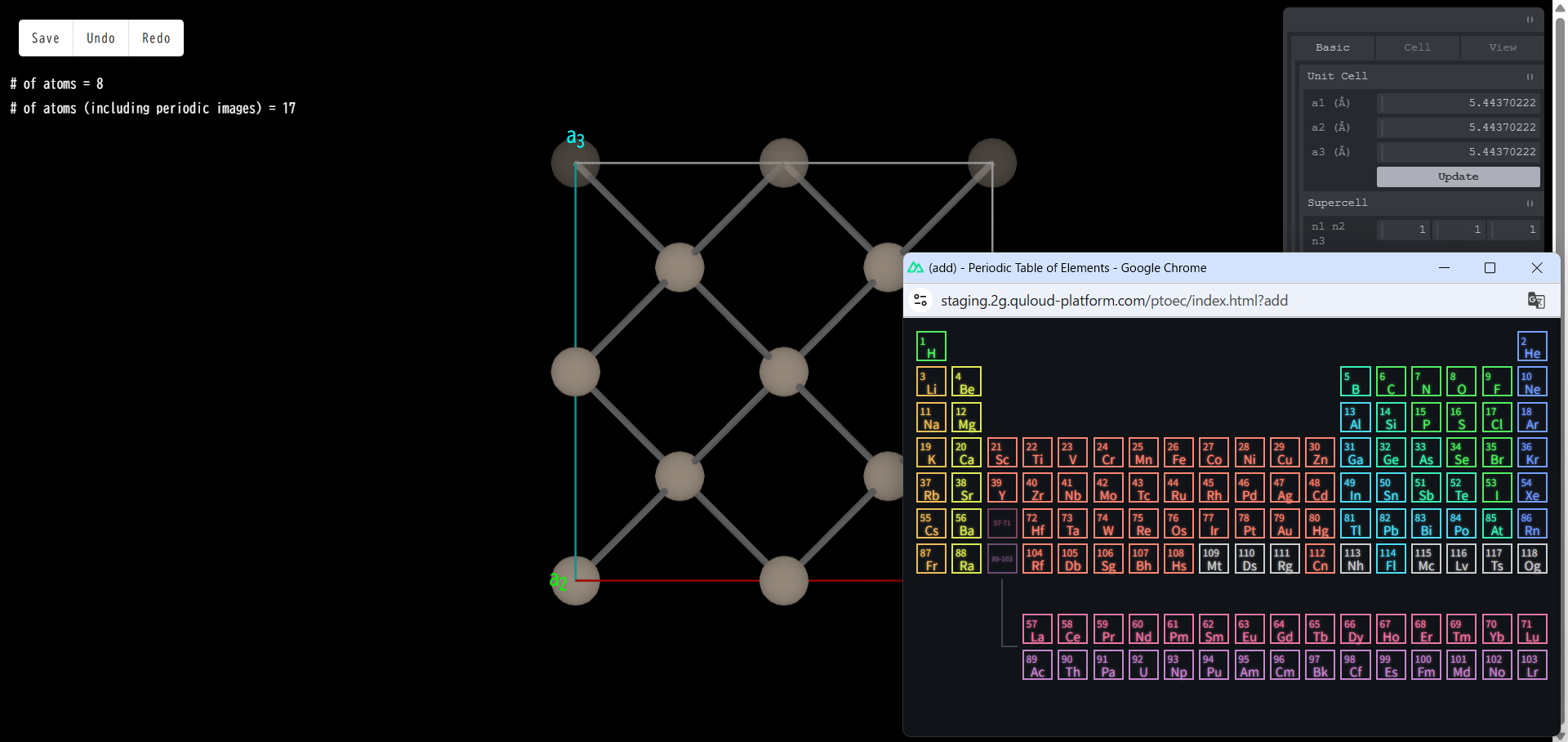
例として「Zn」をクリックすると、本当に Zn 原子を追加するかどうかの確認画面が出ます。
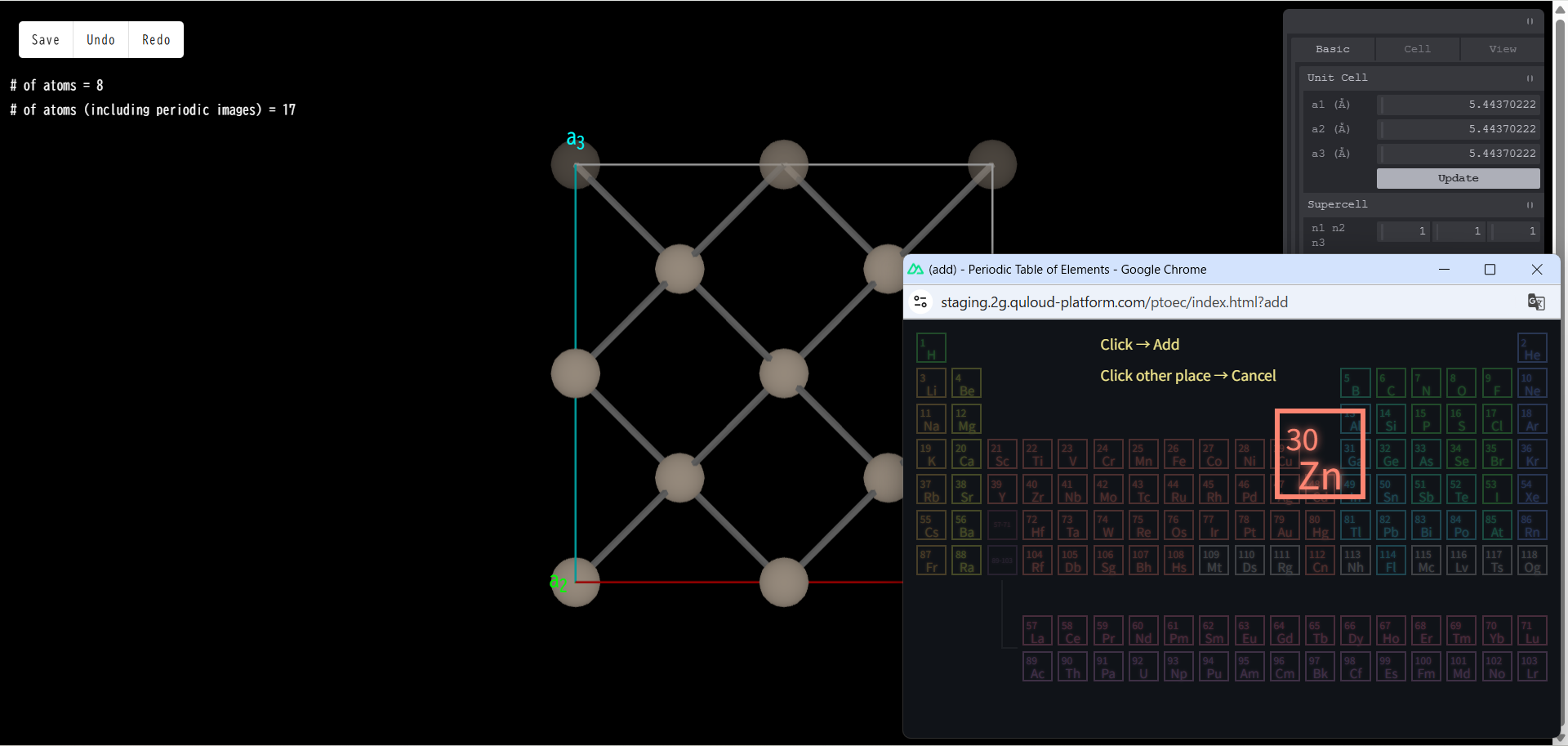
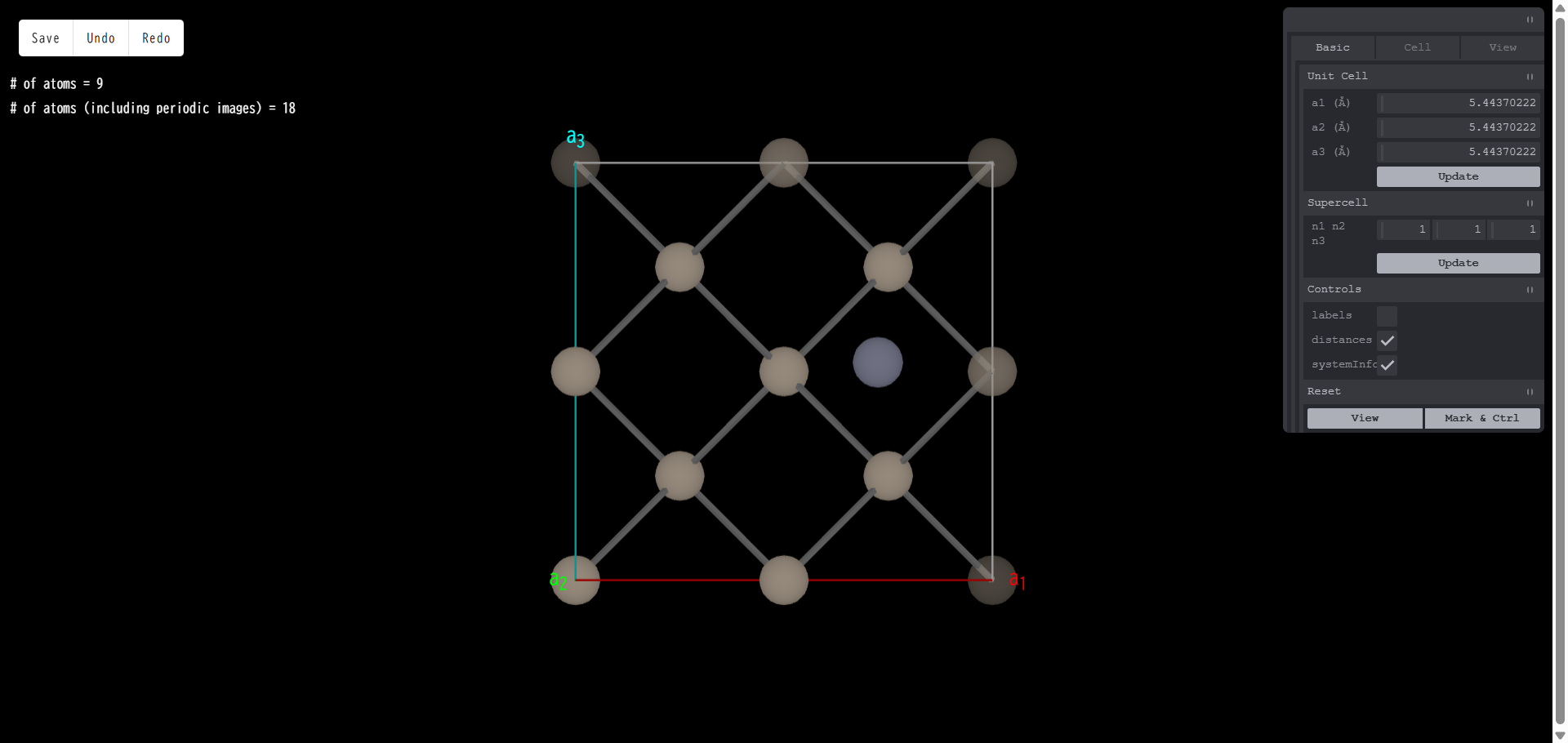
もう一度「Zn」をクリックすると、モデルに Zn 原子が追加されます。
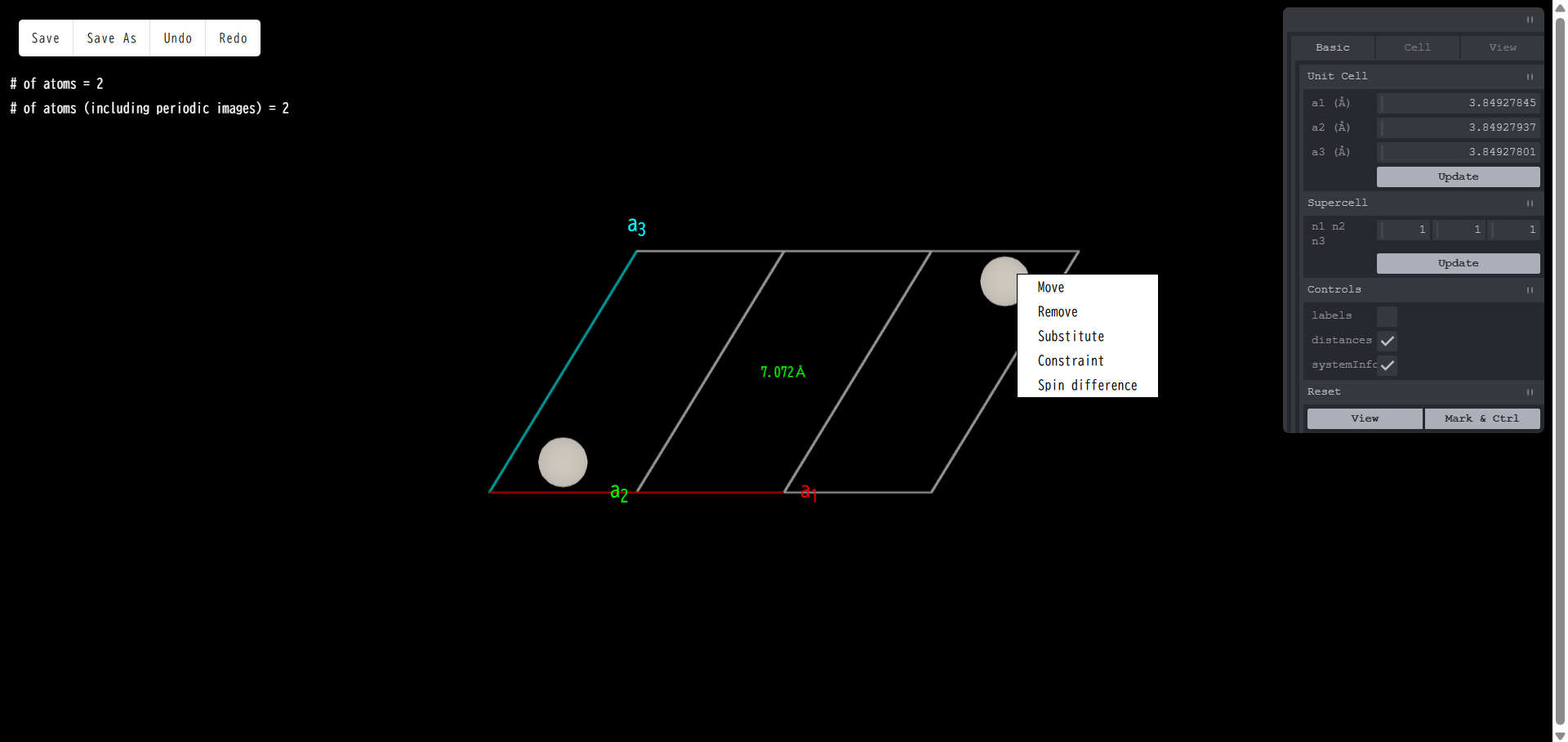
原子の上にマウスカーソルを合わせて右クリックを行った場合は下図のように、 「Move」「Remove」「Substitute」というメニューが現れます。
「移動 (Move)」を選択すると、選択した原子の上に赤、青、緑の3本の矢印が表示されます。
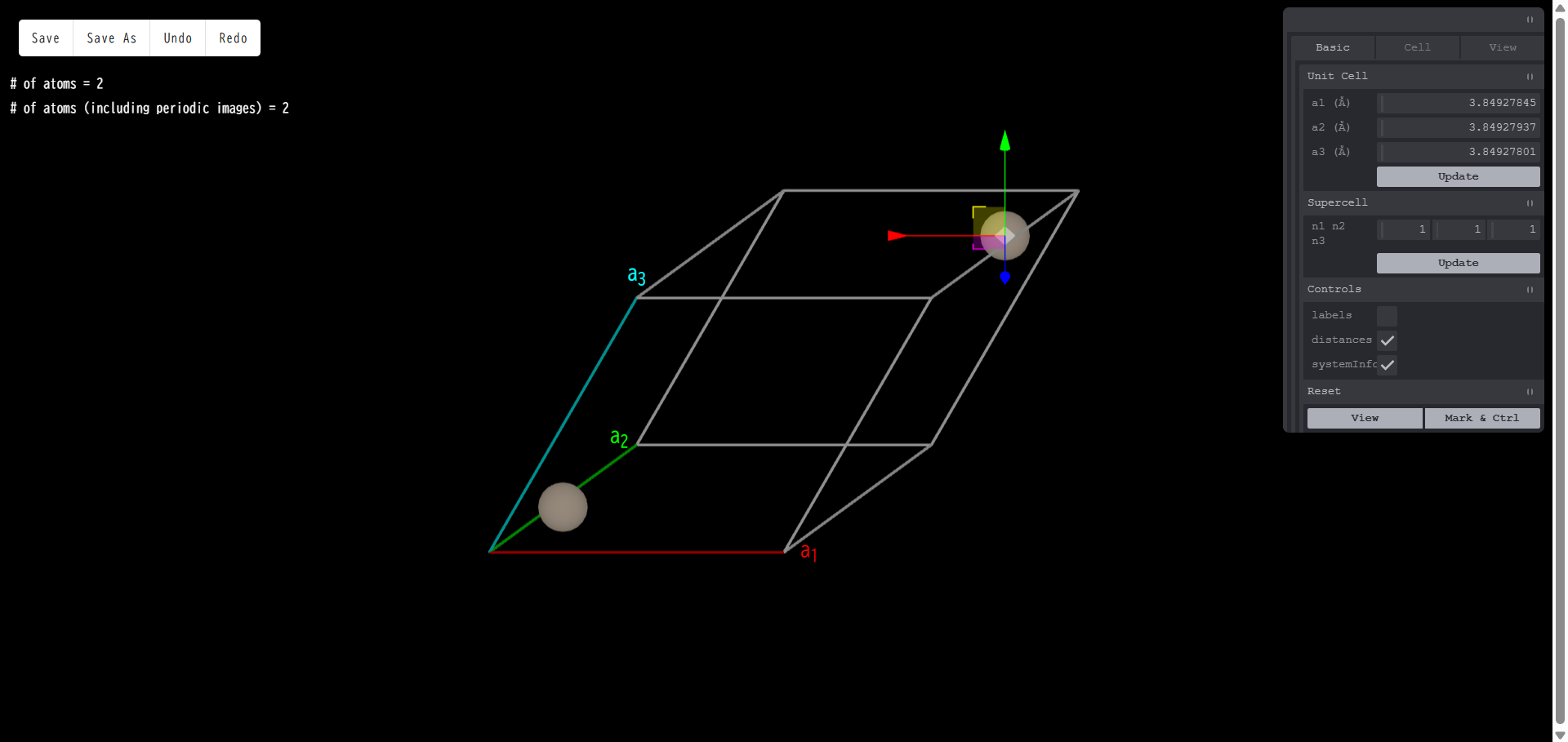
いずれかの矢印をドラッグすると、その矢印の方向に原子を動かすことができます。
また、原子をドラッグすると、方向に関係なく自由に原子を動かすことができます。
「削除 (Remove)」を選択した場合は、その原子がモデルから消えます。
「置換 (Substitute)」を選択した場合は、「Add atom」と同様に、新しいウィンドウで元素周期表が現れます。 ここで、どの元素の原子で置換するか選択します。
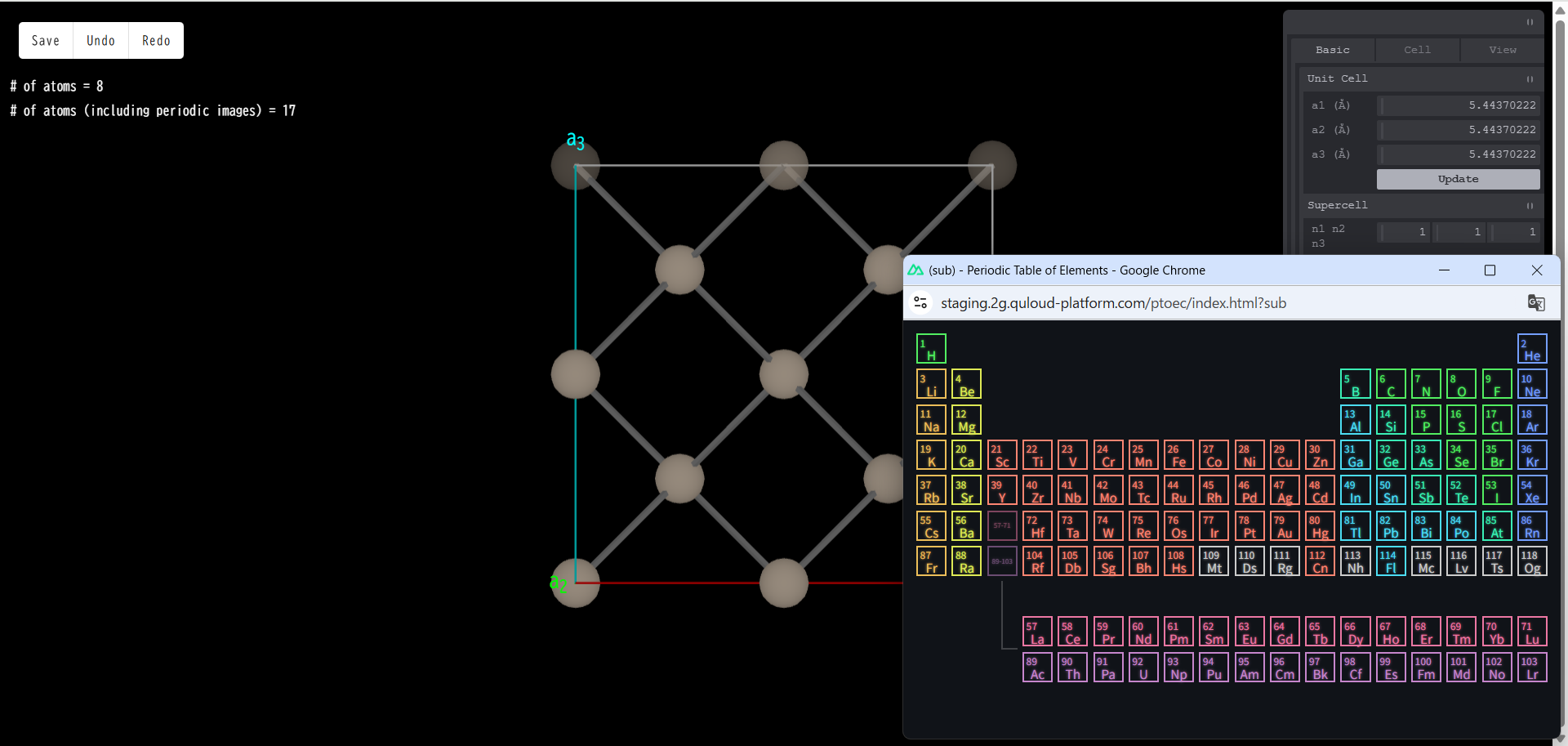
例として「Fe」をクリックすると、本当に Fe 原子で置換するかどうかの確認画面が出ます。
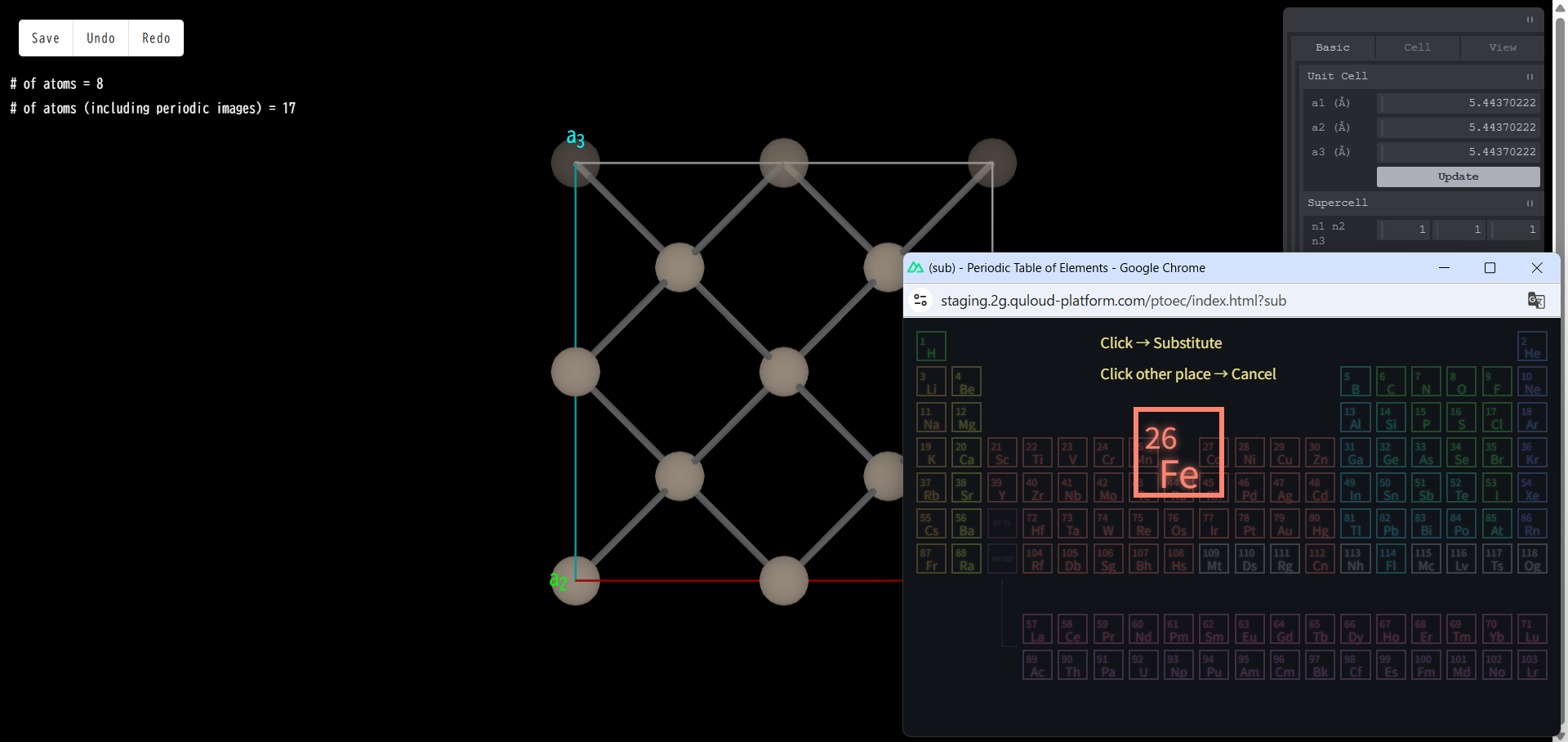
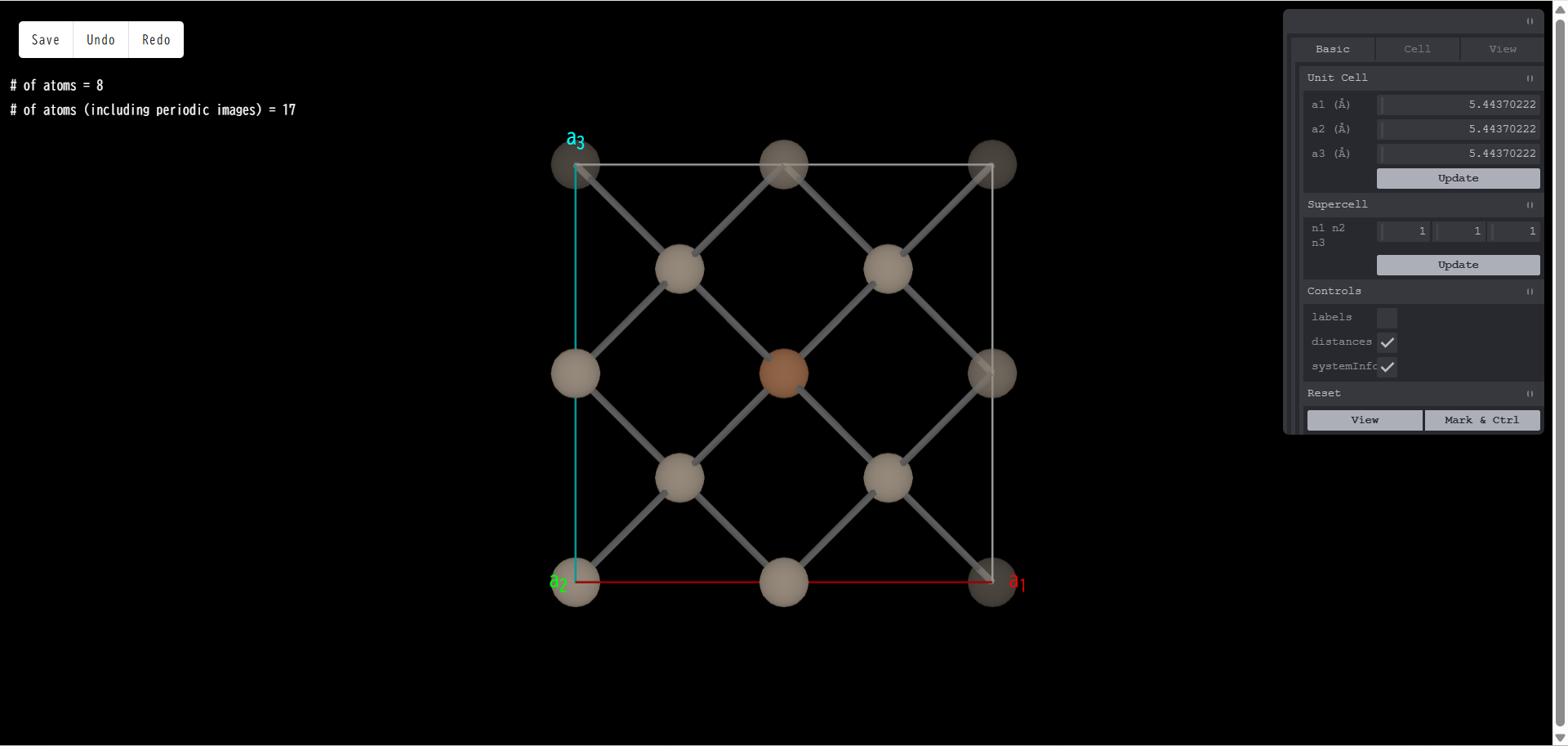
もう一度「Fe」をクリックすると、 Fe 原子で置換されます。
7.2. モデリングのやり直しと中止
ブラウザのリロードを行うと、全ての操作はリセットされ、モデリング開始時点の状態に戻ります。 また、モデリングのタブを閉じると、モデリングそのものを中止して、元の Quloud の利用に戻る事ができます。
7.3. 操作パネル
モデリングの主な操作は右側パネルのメニューから行います。 また、もう1つの別の Material を組み合わせてモデリングを行う場合の補助的な役割を、 フッターメニューが担います。
Material が分子の場合は下図のように操作パネルの内容が少し異なります。

まずは Material が結晶の場合のモデリングについて説明します。
7.4. Lattice Parameters
Lattice Parameters メニューを開くと、現在表示されている Material の格子の長さと角度の値を変更するフォームが現れます。 設定したい値を入力し、最後に「Apply Cell Parameter Change」ボタンをクリックすると、例えば下図のような構造が得られます。
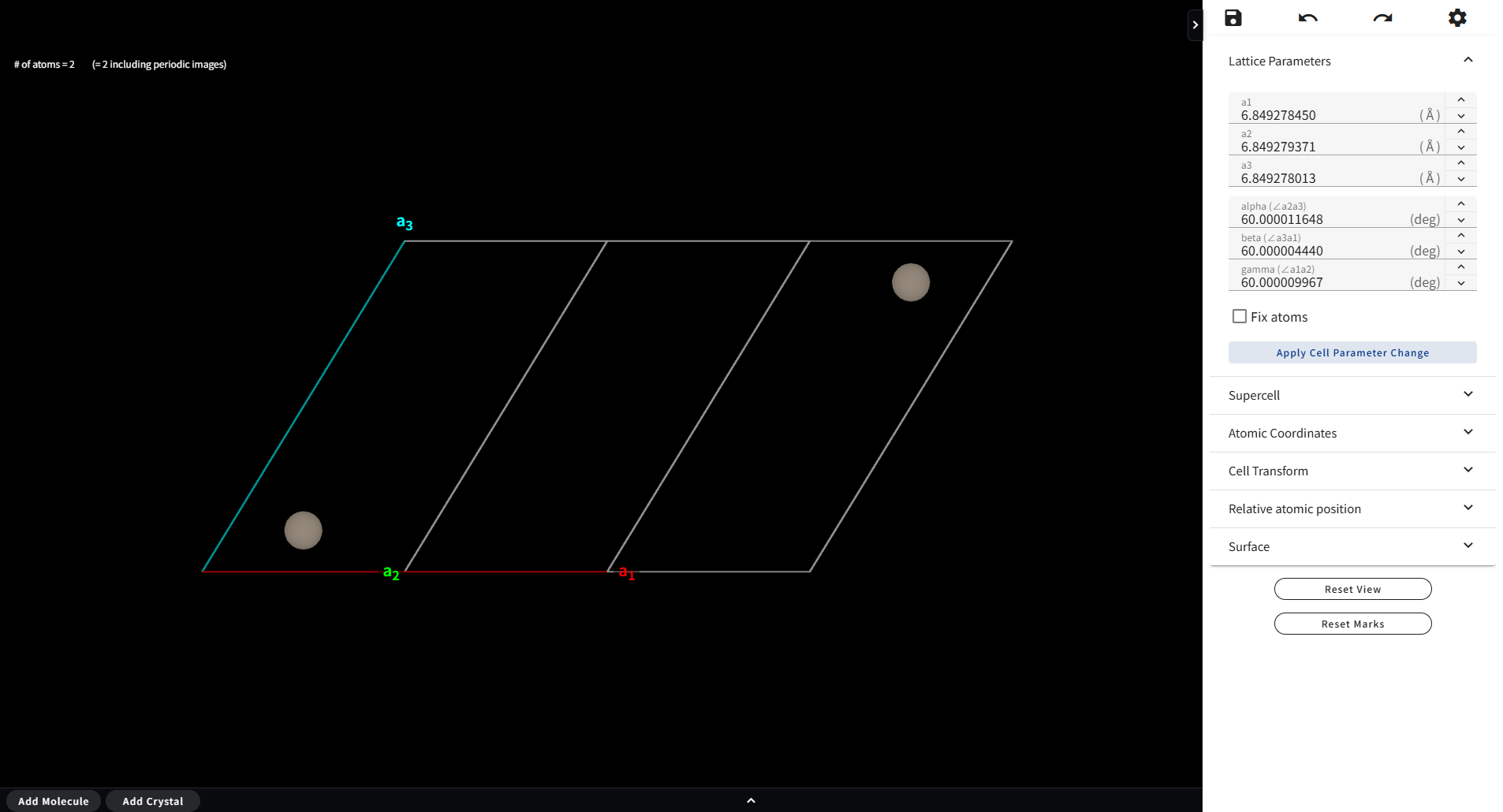
通常は、上図のように、原子位置も格子のサイズ比例して変化しますが、 「Fix atoms」というチェックボックスにチェックを入れて格子定数を変えると、 原子位置は固定したまま、格子のサイズだけを変えることができます。
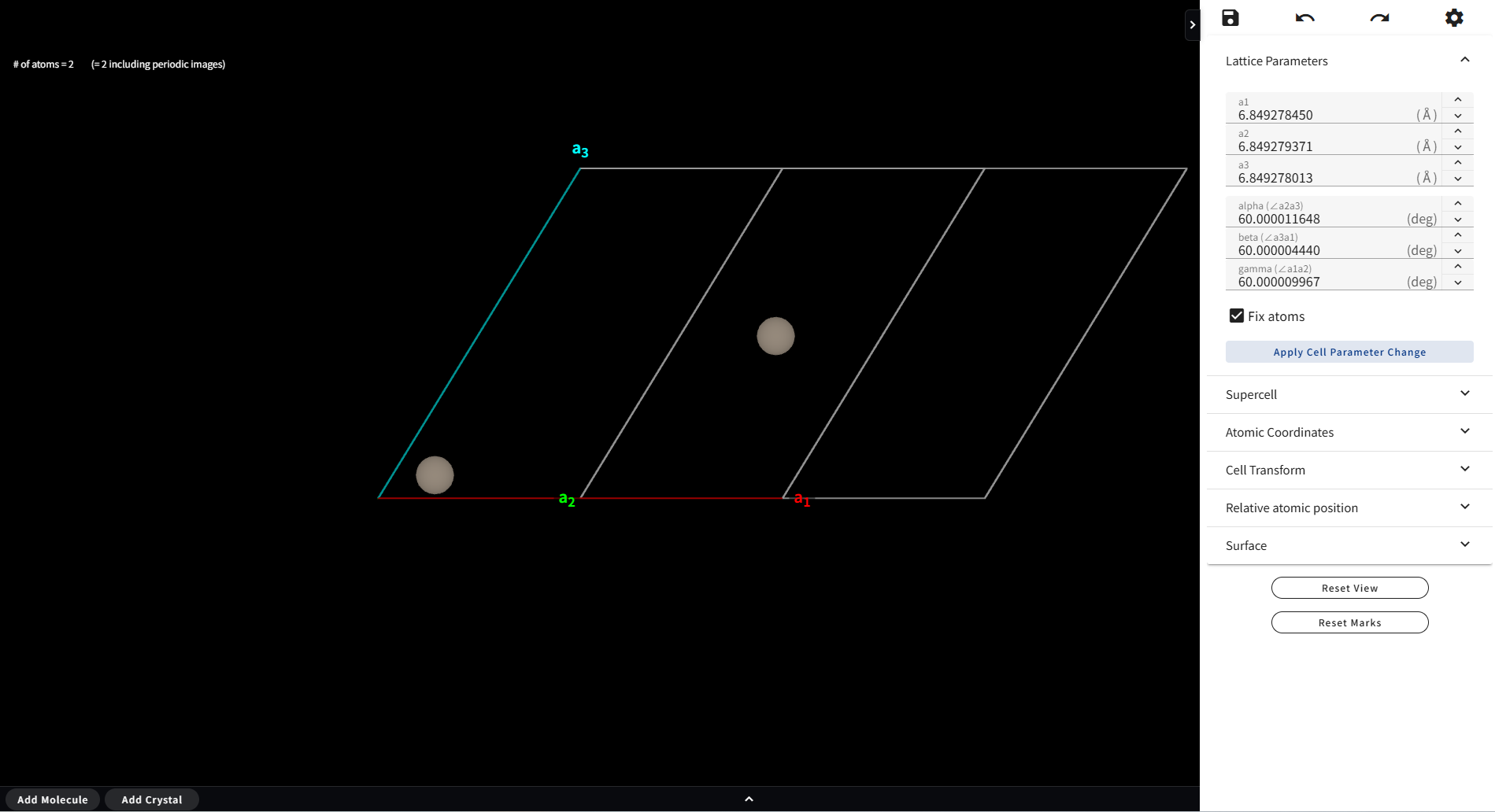
Tip
Fix atoms を利用すれば、表面スラブモデル等で真空領域のサイズを調整することができます。
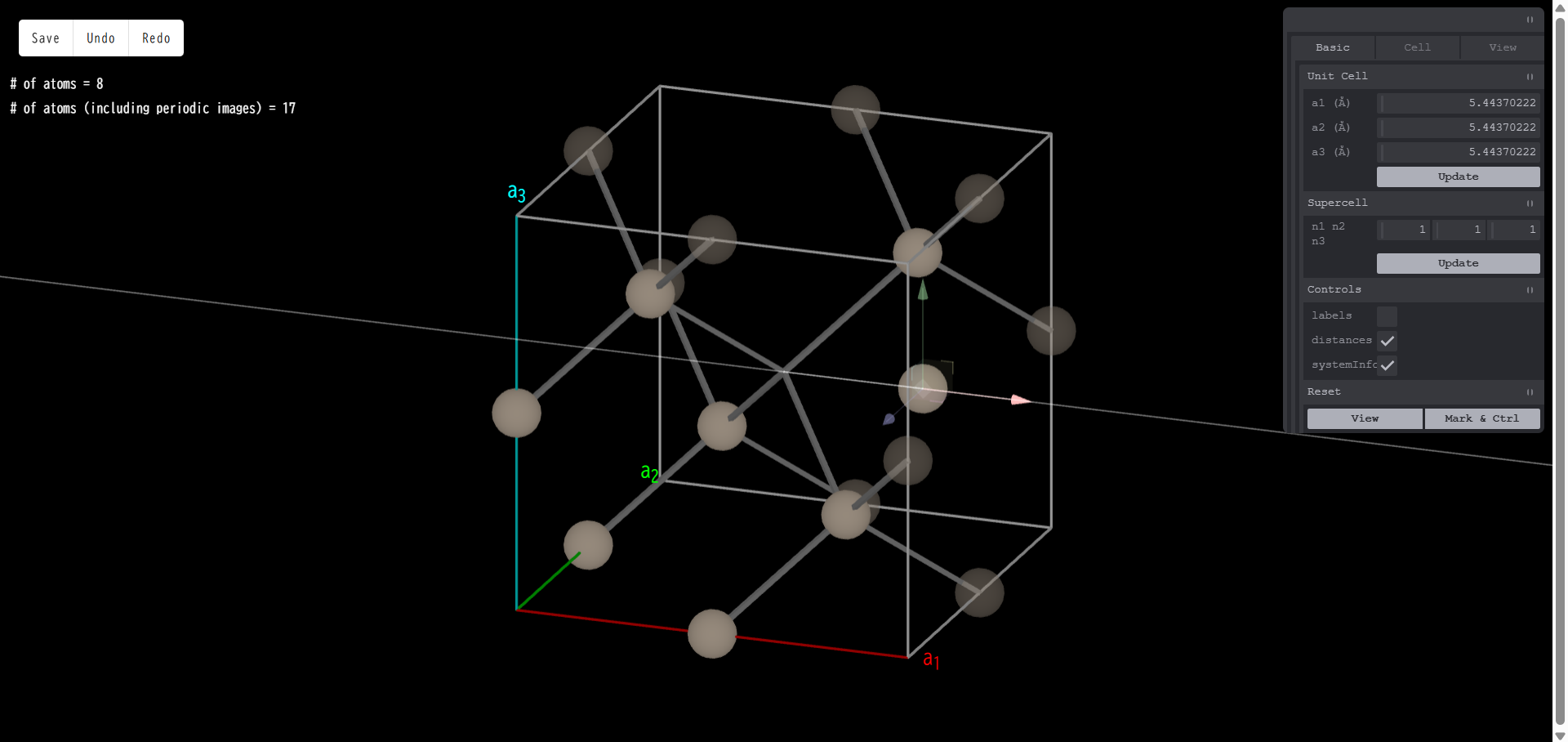
7.5. Supercell
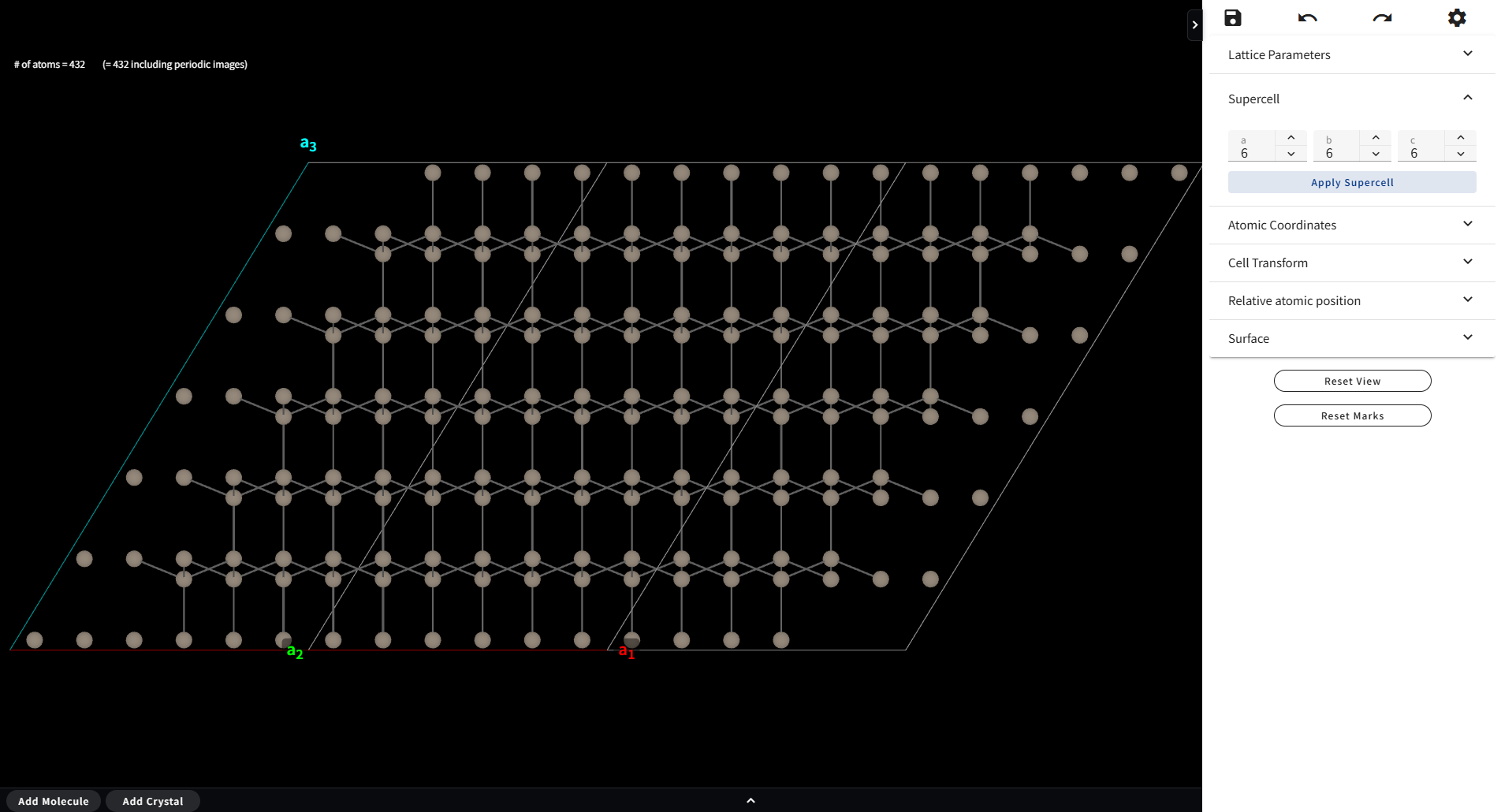
Supercell メニューを開くと、3つの整数値を入力するフォームが現れます。 値を設定し、「Apply Supercell」ボタンをクリックすると、下図のようなスーパーセルが作られます。
7.6. Atomic Coordinates
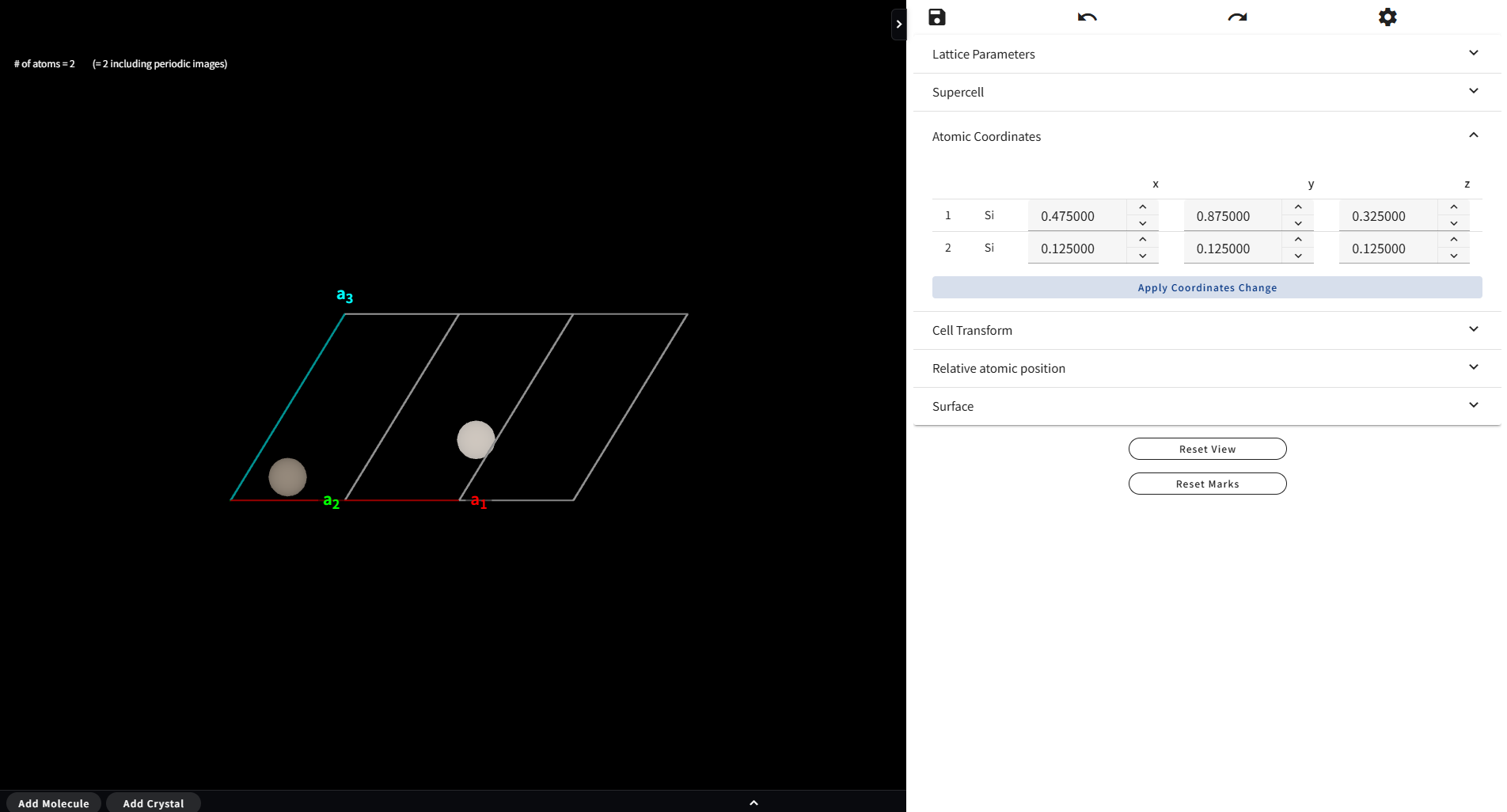
Atomic Coordinates メニューを開くと、個々の原子の座標を入力できるフォームが現れます。 値を設定し、「Apply Coordinates Change」ボタンをクリックすると、設定した座標に対応した位置に原子が移動します。
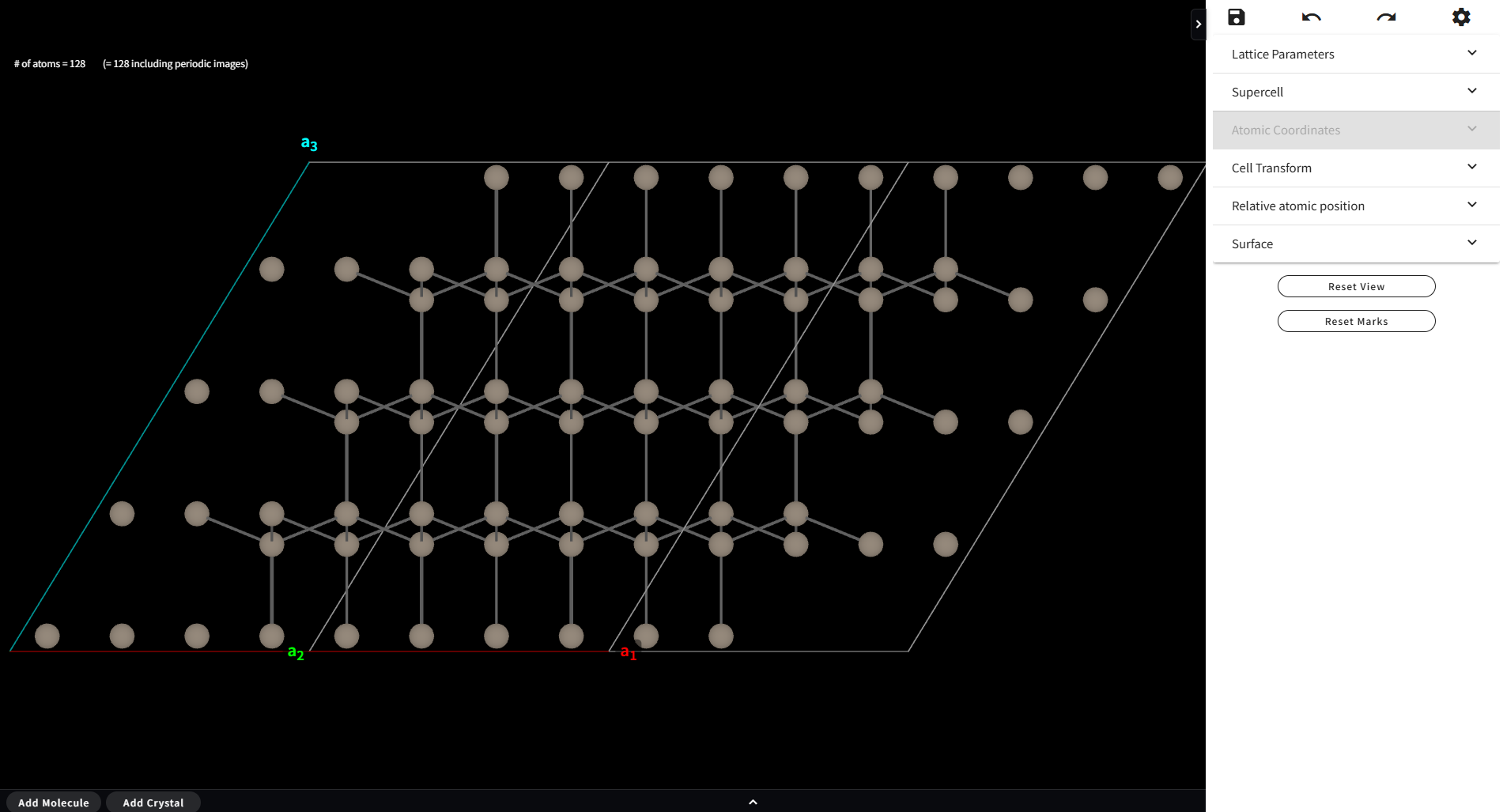
ただし、原子数が多い Material(>100原子)では、このメニューは利用不可になります(下図)。
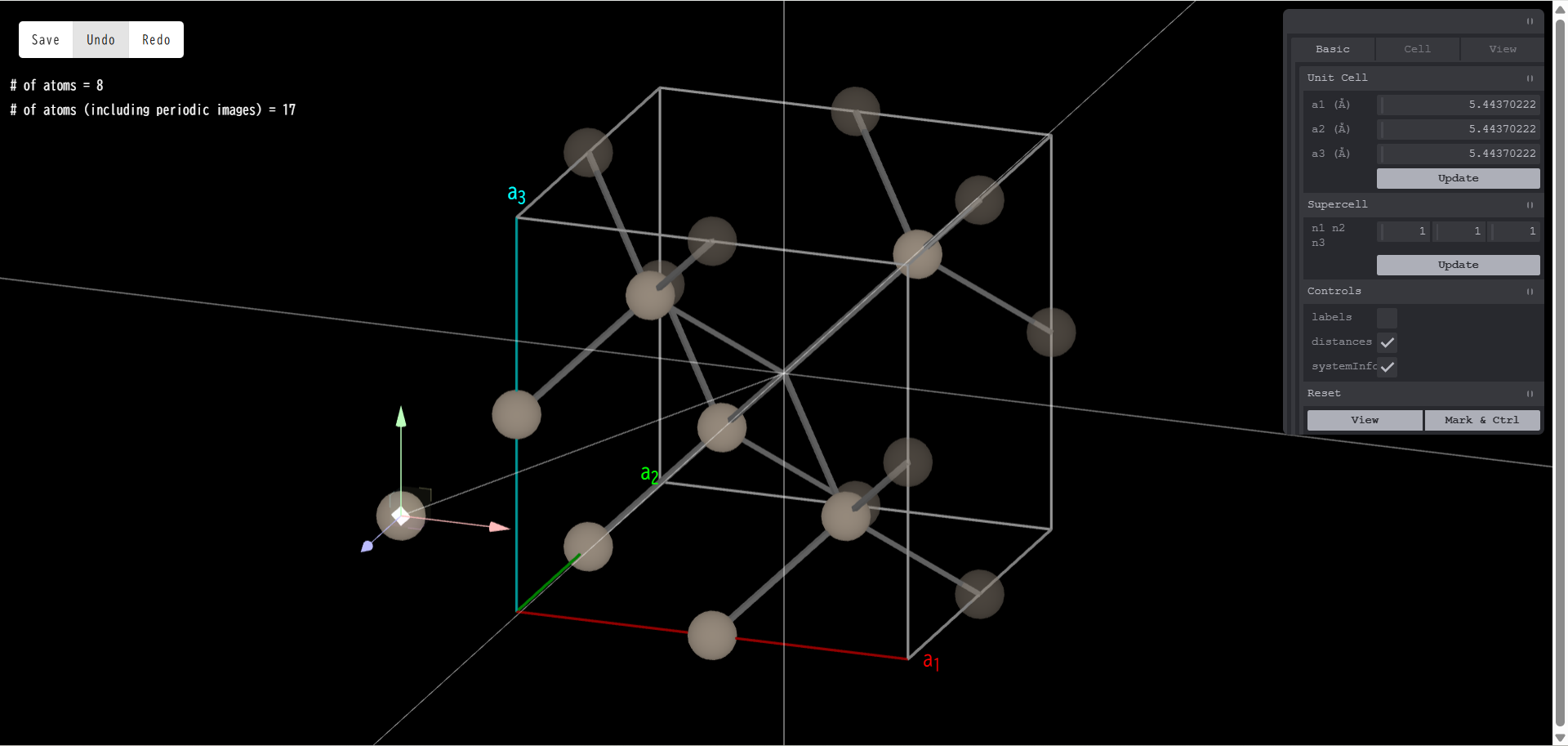
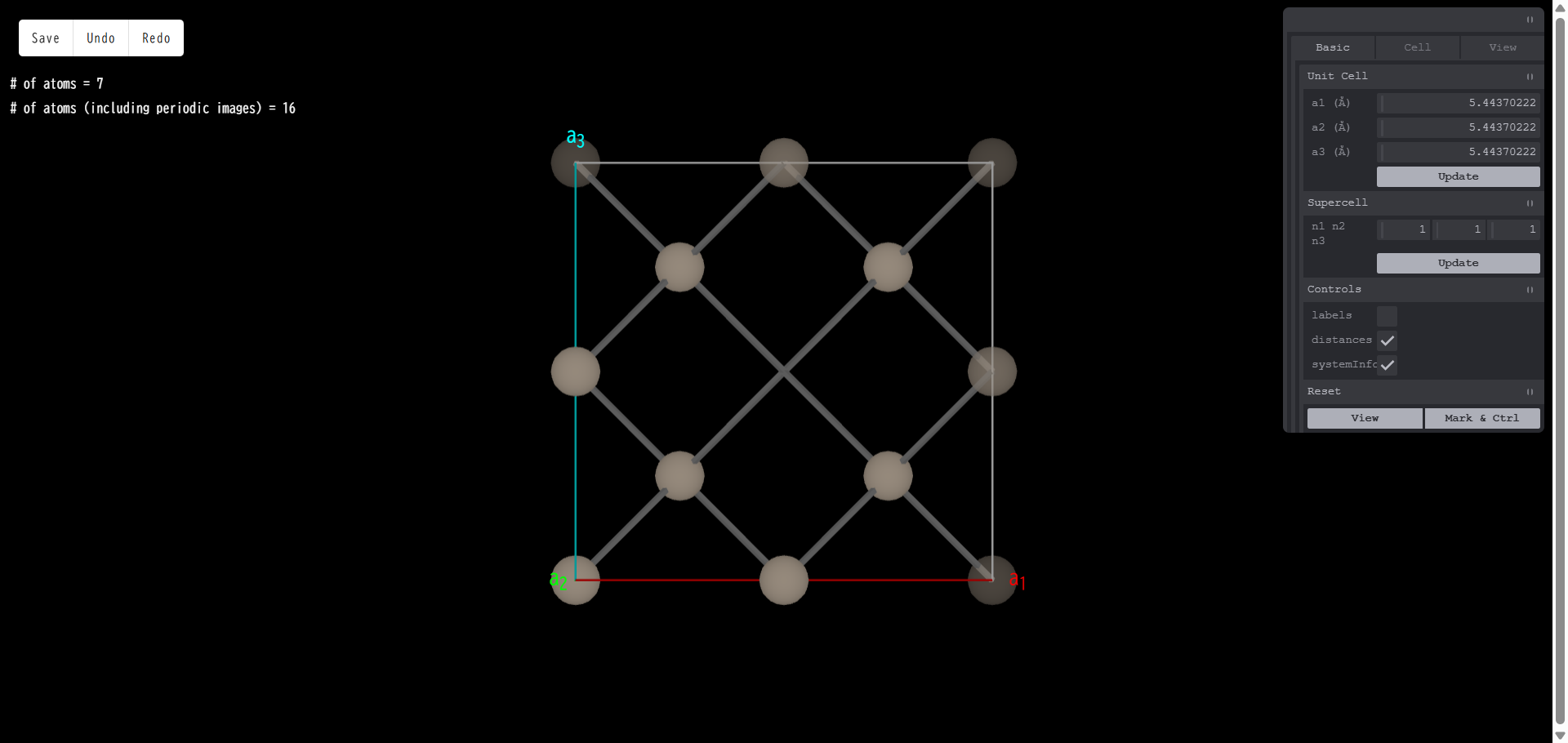
7.7. Cell Transform
Cell Transform メニューを開くと、「Primitive」「Conventional」のボタンがあります。 クリックすると(系の対称性が上手く読み取れれば)、 その系の最小単位胞(Primitive Cell)あるいは慣用単位胞(Conventional Cell)を作る事ができます (両者が同じになることもあります)。 下図は、bcc 構造の Fe の単位胞(Primitive でも Conventional でもない)です。
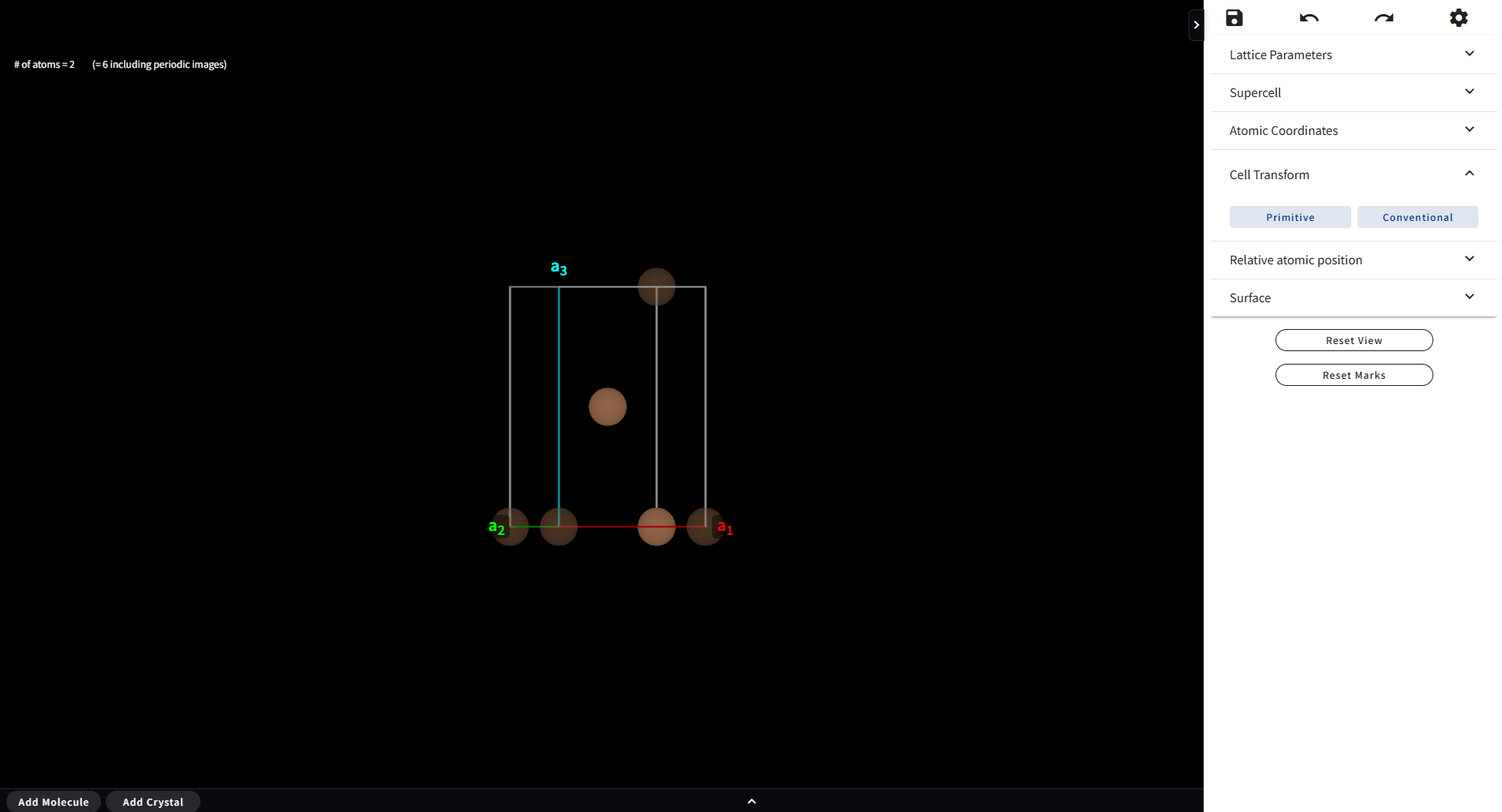
これを Primitive Cell に変換すると下図のようになり、
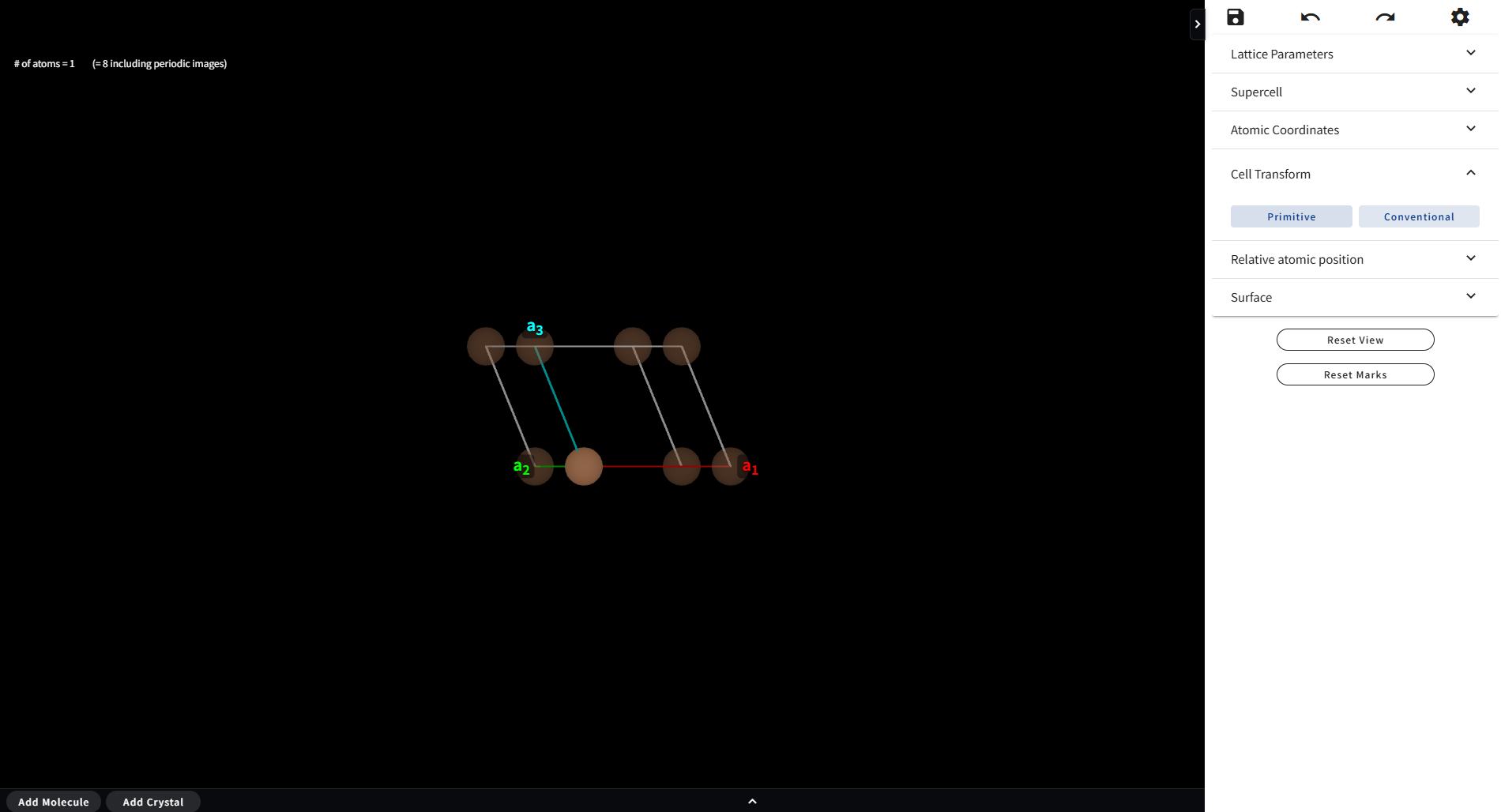
Conventional Cell に変換すると下図のようになります。
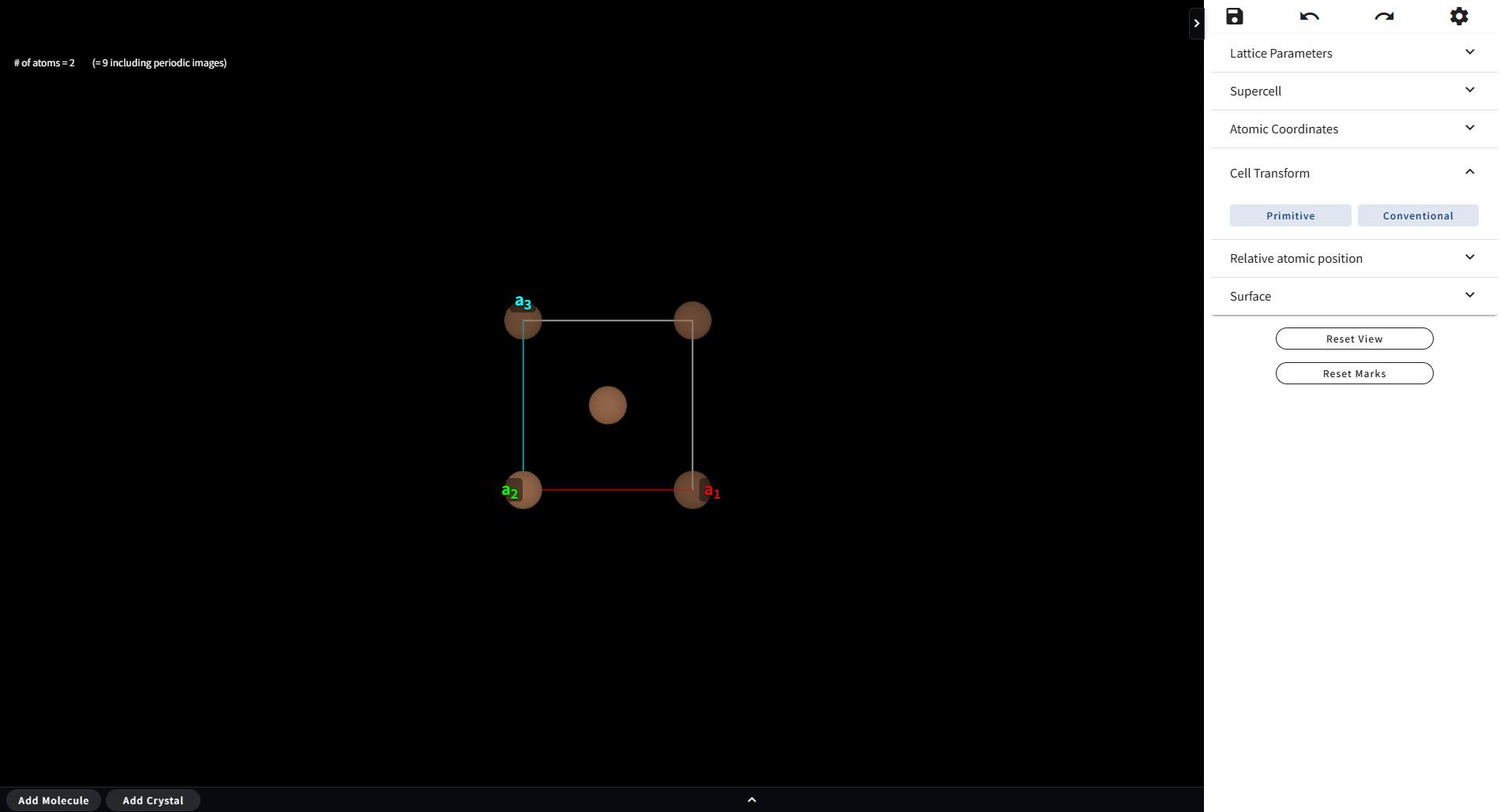
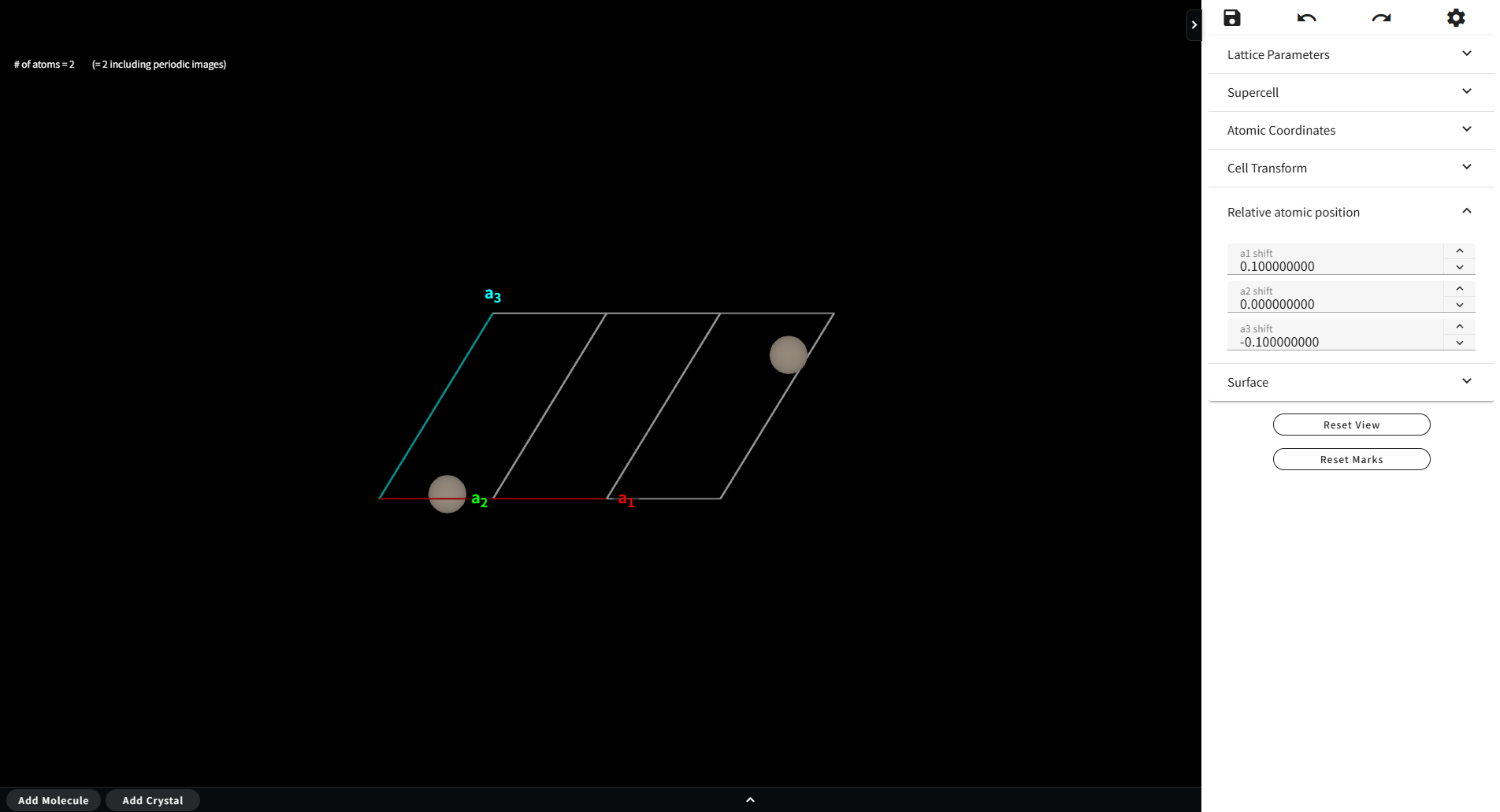
7.8. Relative Atomic Position
Relative Atomic Positoin のメニューを開くと、3つの数字を入力するフォームが現れます。 この数字に応じて、系内の原子全体が(対応する格子ベクトルの方向に)平行移動します。 取り得る範囲は -1 ~ +1 です(格子ベクトルの長さ分だけ ± 方向に移動できる)。
7.9. Surface
Surface メニューには、表面スラブと呼ばれる下図のような原子構造を作成するための設定フォームがあります。
設定パラメータは以下の通りです。
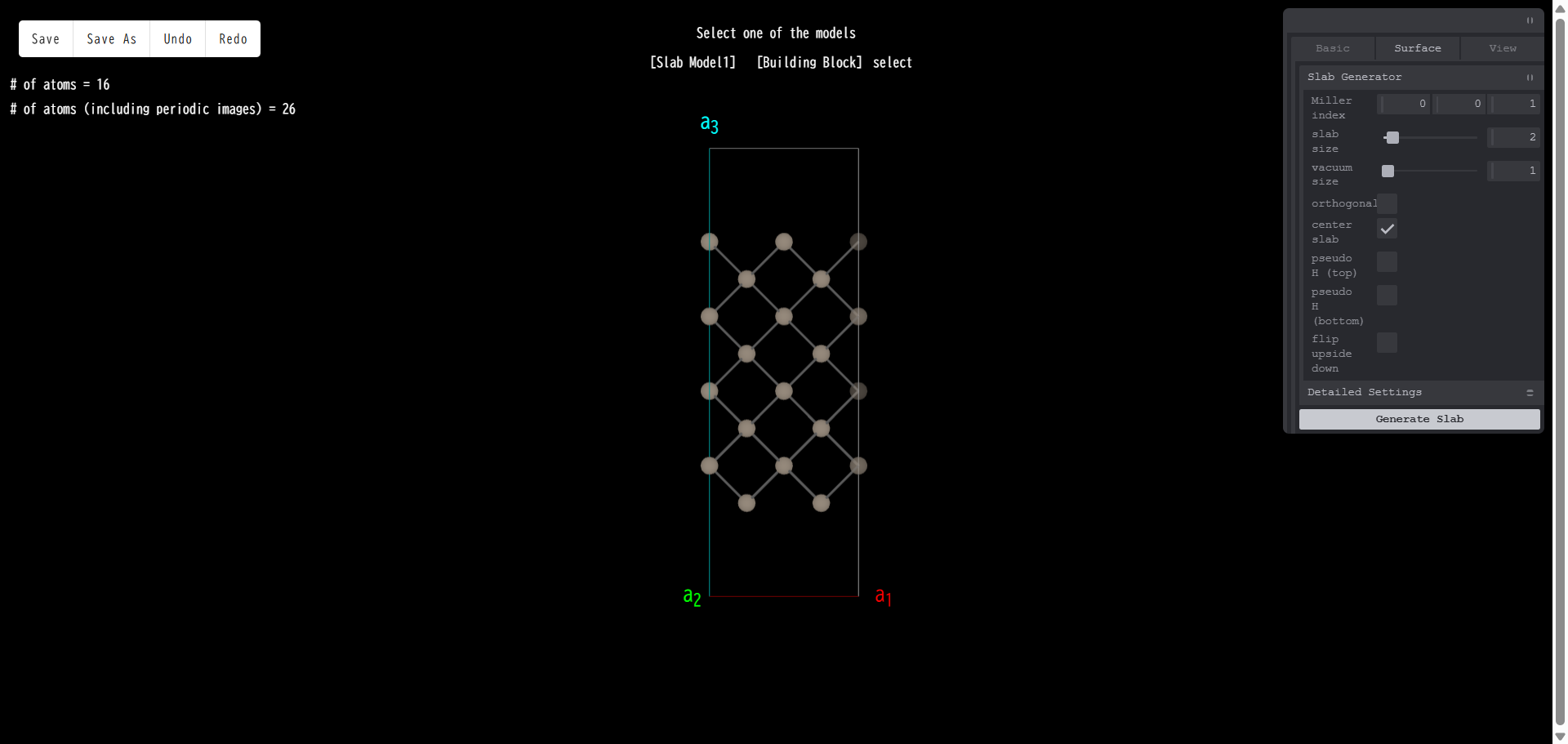
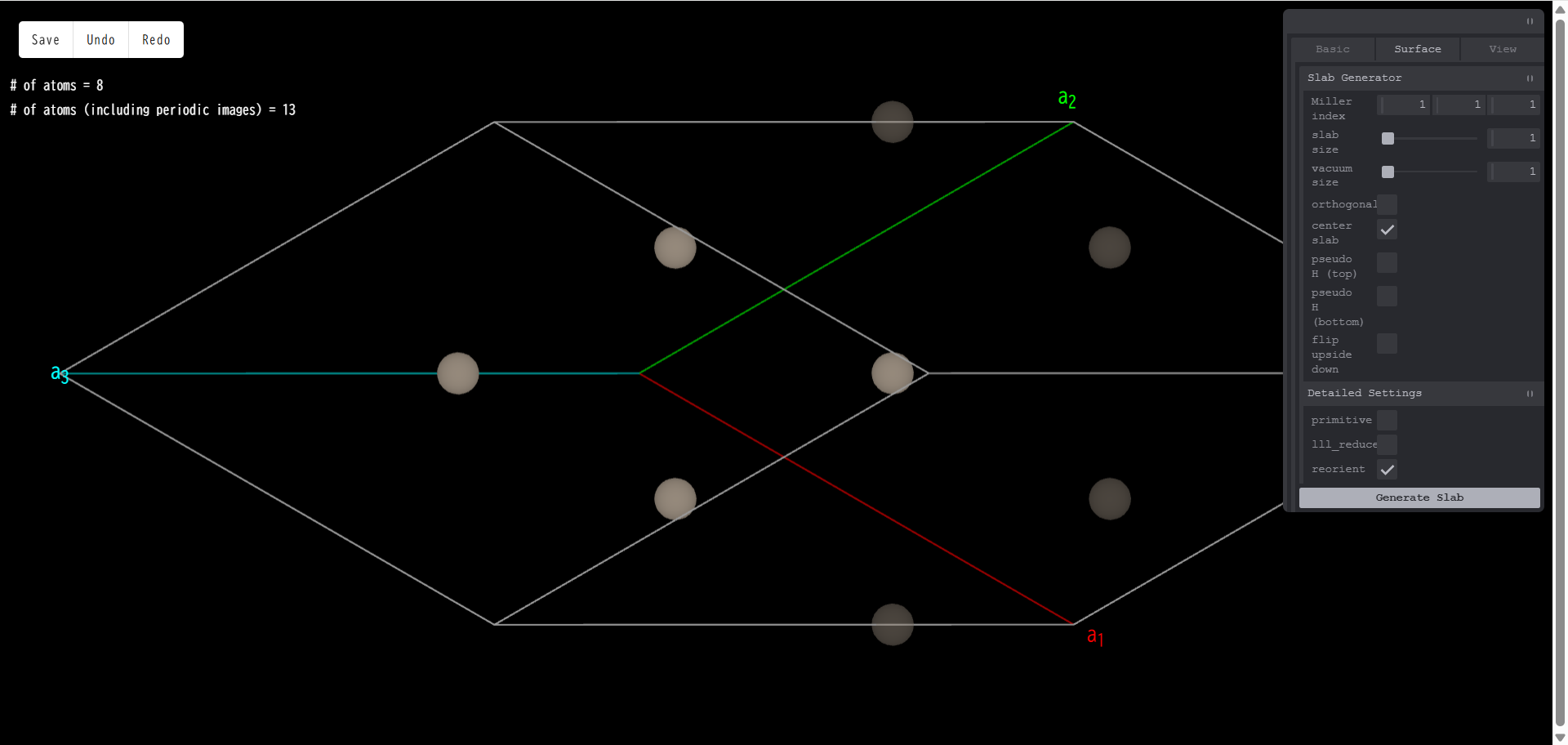
Miller index:真空領域に露出する表面のミラー指数を指定します。例えば、Si のコンベンショナルセルで表面スラブモデルを作成する際、ミラー指数を (001) とし、「Generate Slab」を押すと、スラブモデルの側面図は次のようになります。
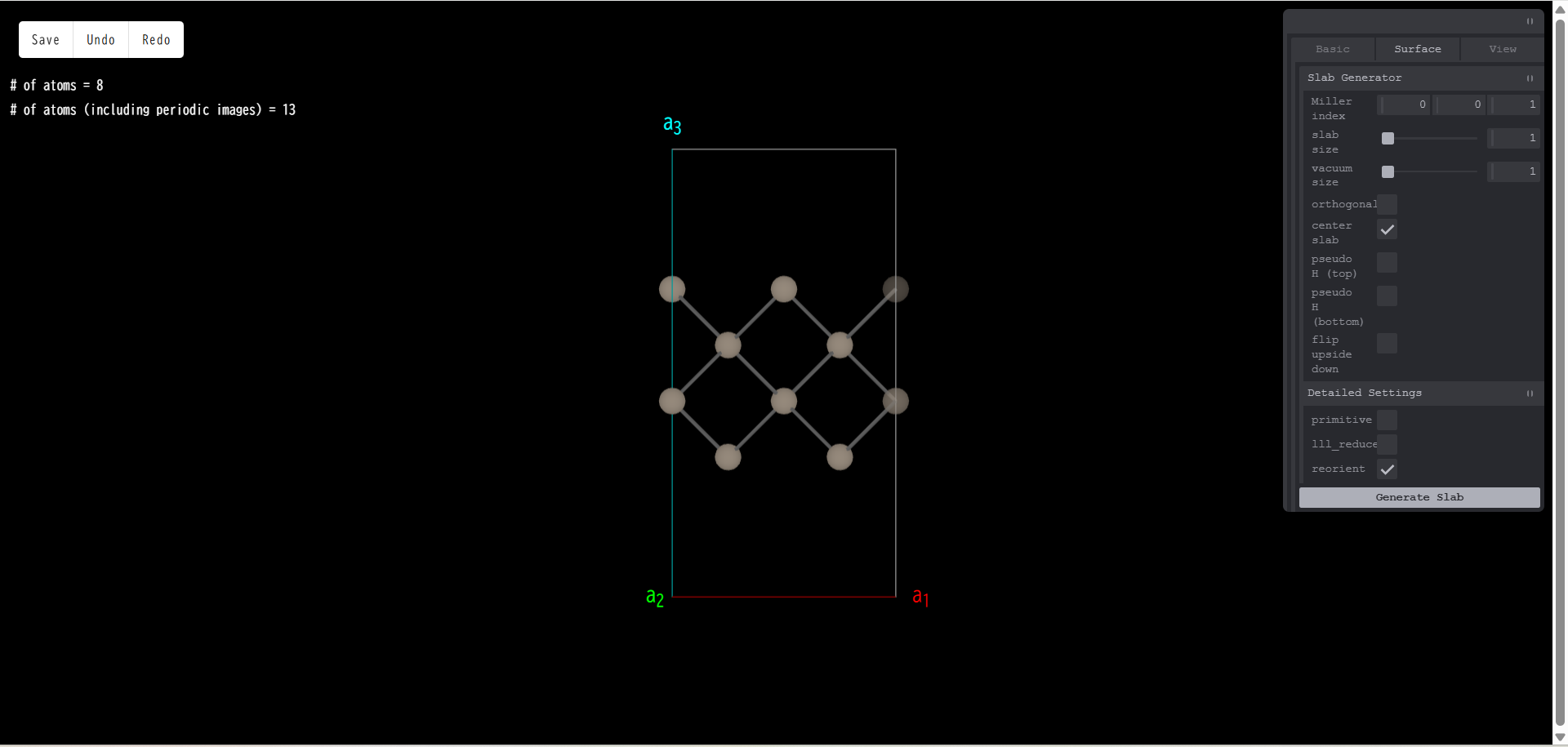
上面図は次のようになります。
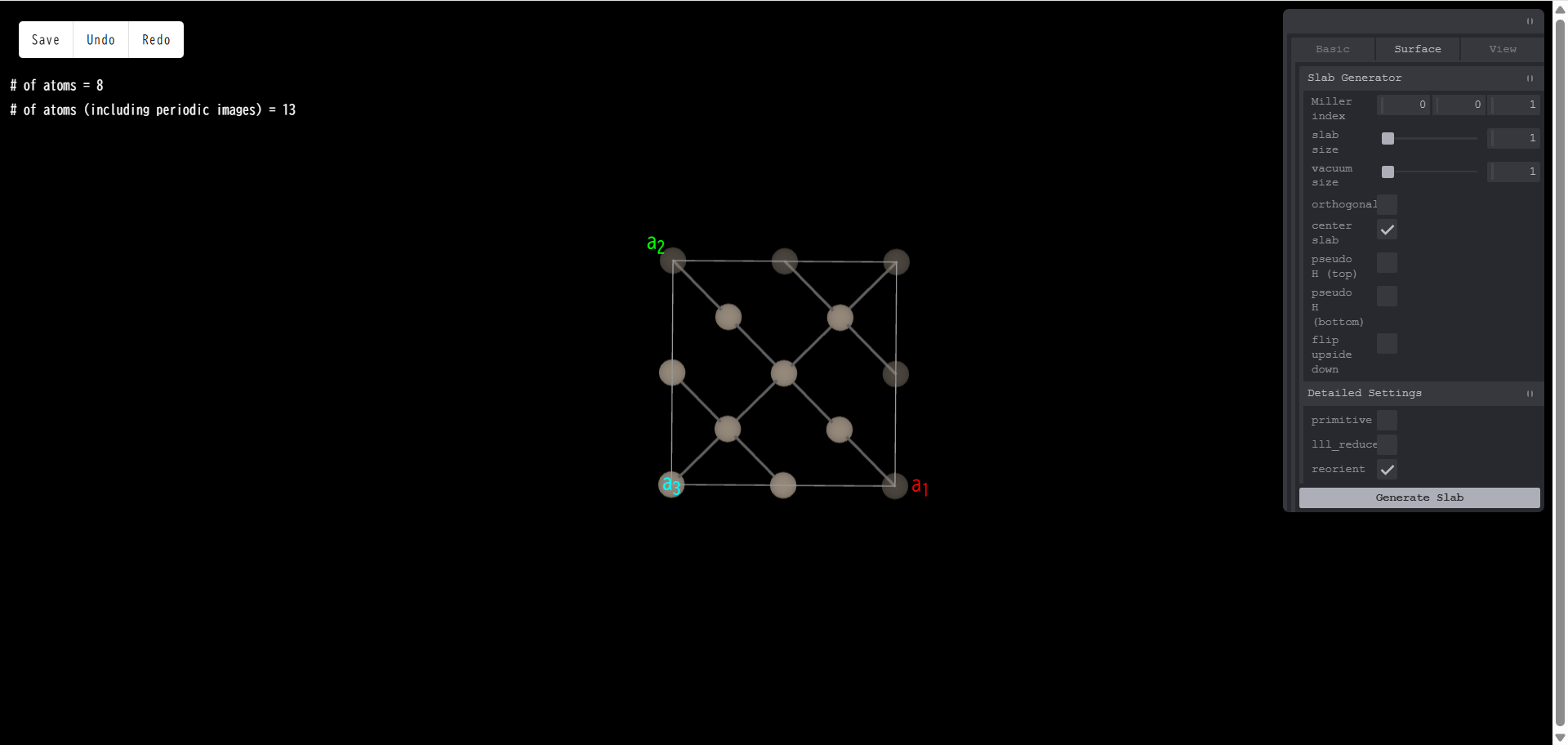
一方で、ミラー指数を (111) とし、「Generate Slab」を押すと、真空領域に露出する表面が変わり、側面図は次のようになります。
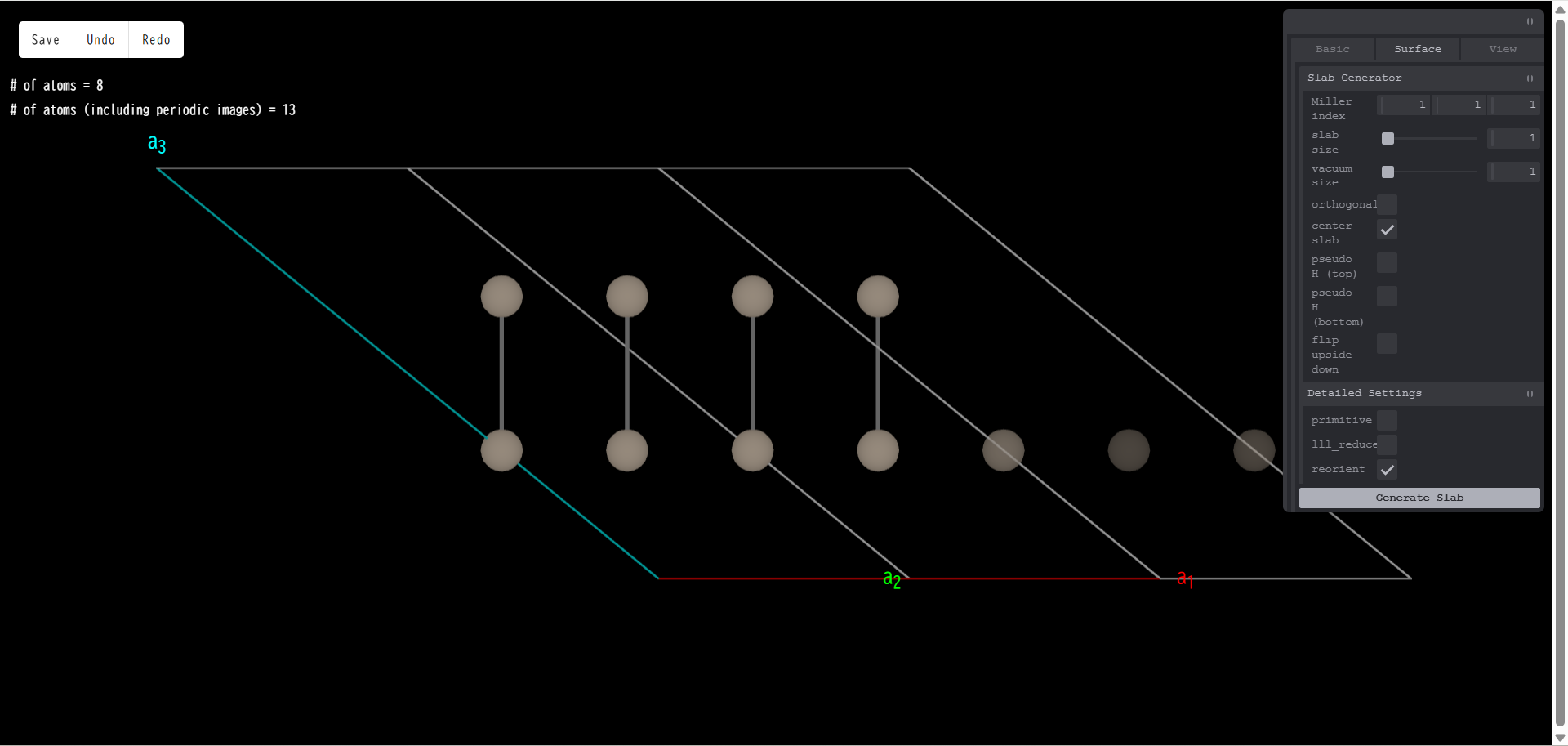
上面図は次のようになります。
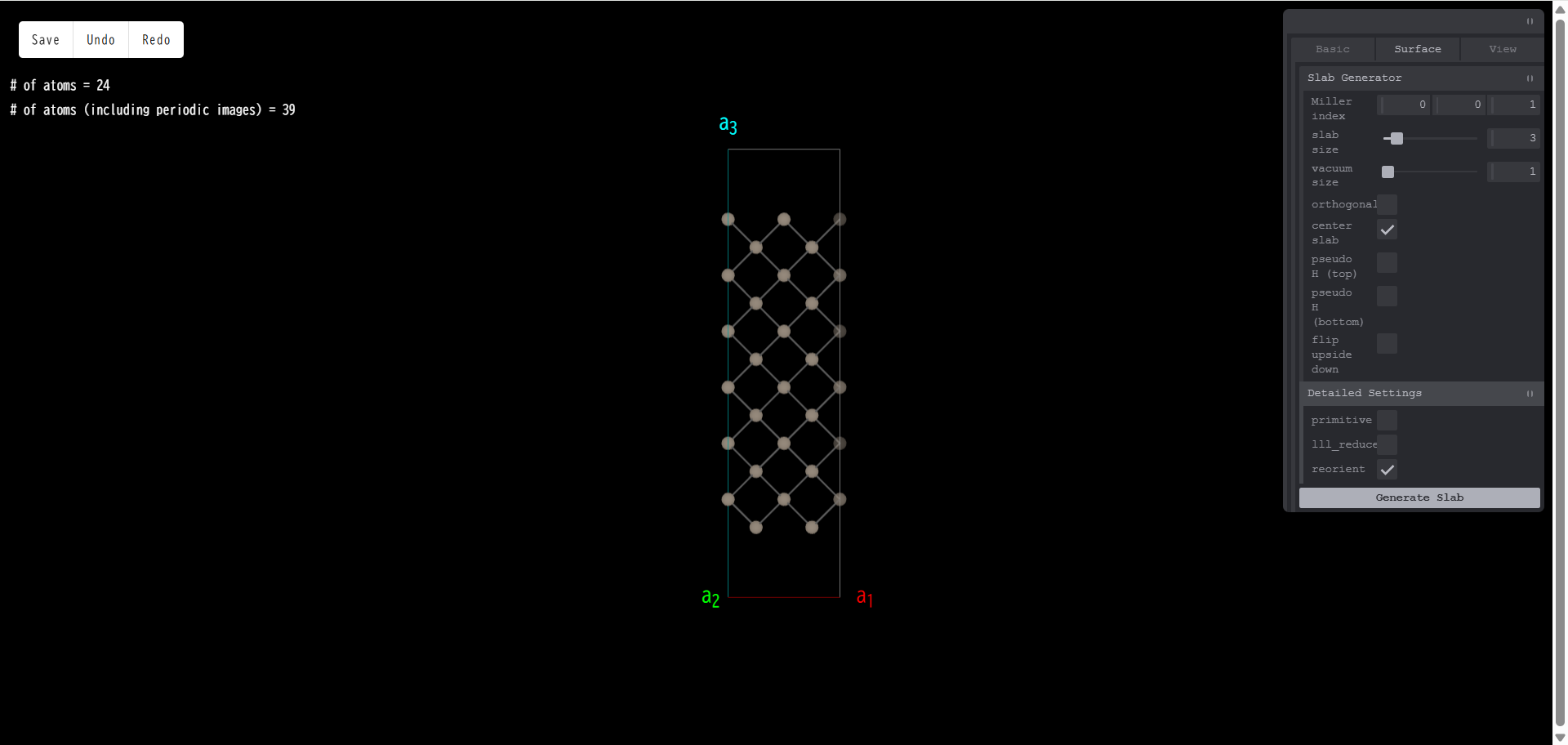
slab size:スラブ層の厚さを指定します。元になるバルク構造のユニットセルを単位とします。例えば、スラブ層厚さを3とし、「Generate Slab」を押すと、次のようになります。
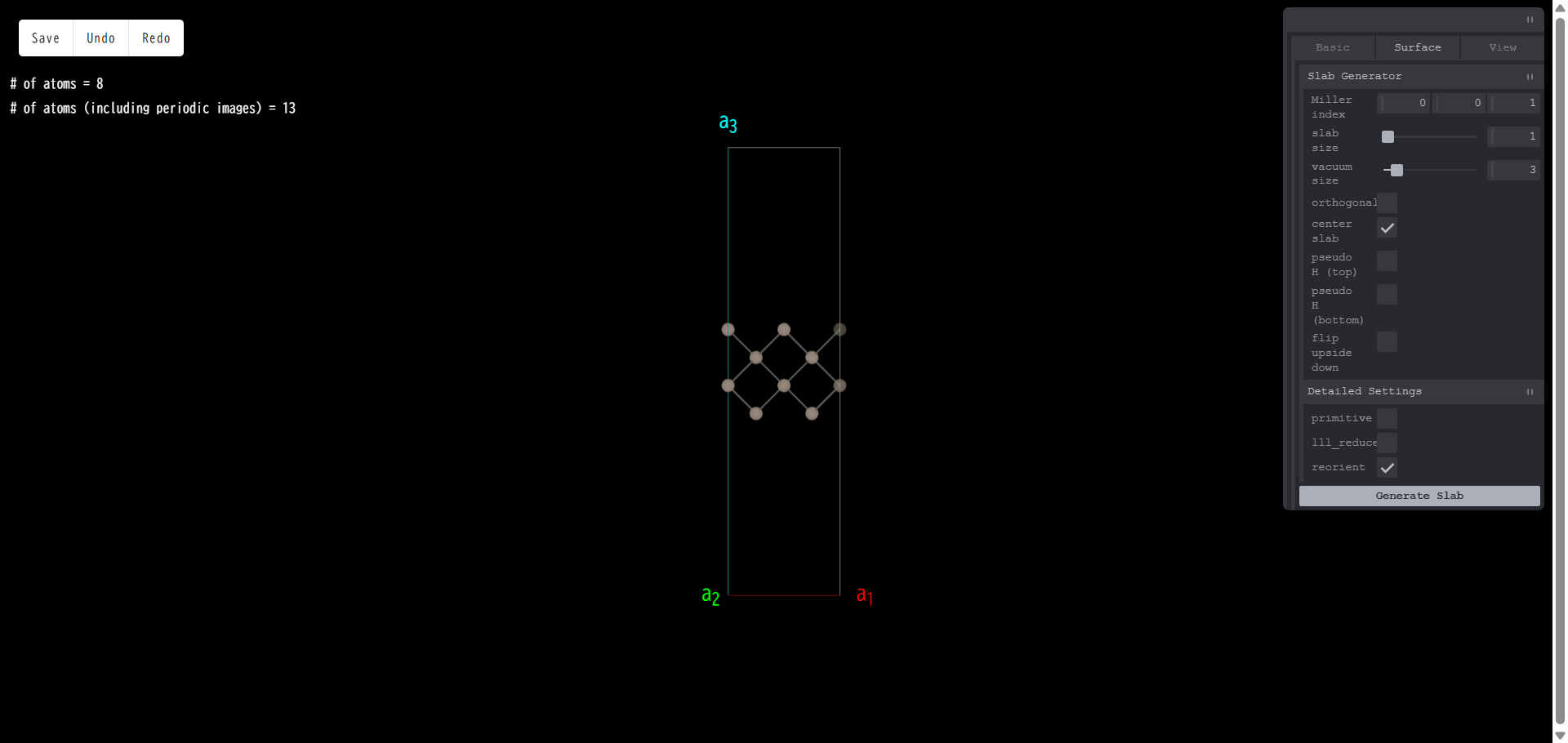
vacuum size:真空層の厚さを指定します。単位はスラブサイズと同じです。例えば、真空層厚さを3とし、「Generate Slab」を押すと、次のようになります。
orthogonal c slab:表面に垂直な軸を c 軸とするセルを取ります。
警告
この機能は、格子ベクトル a3 が表面に対して厳密に垂直になるように強制的にセルを取り直すため、Material 本来の構造からズレが生じる場合があります。
例として、Si の Primitive セルをバルク構造とするスラブモデルでは、下図のように、格子ベクトル a3 は表面に対して垂直にはなりません。
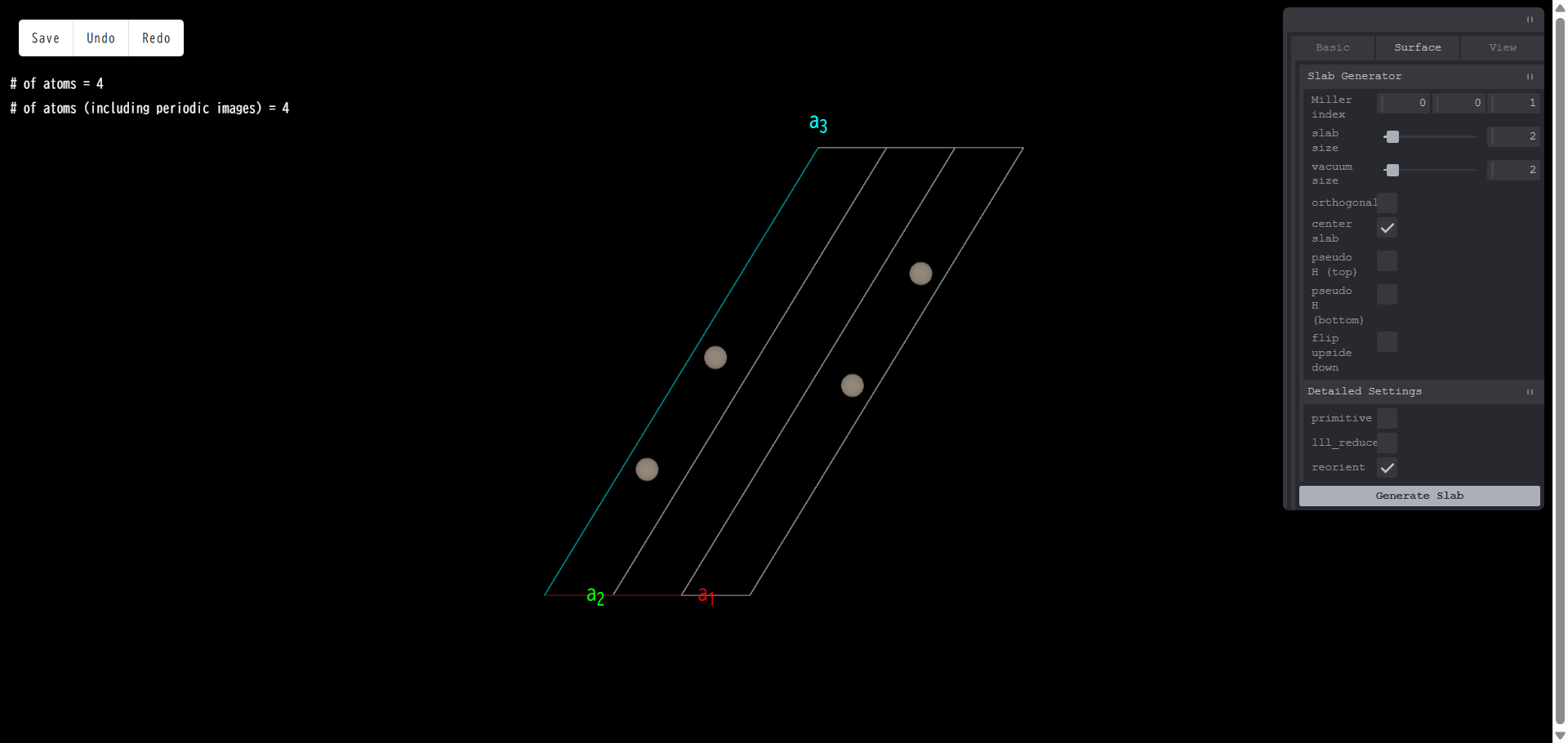
ここで「orthogonal c slab」にチェックを入れ、「Generate Slab」すると、表面に垂直になるように、格子ベクトル a3 が取り直されます。
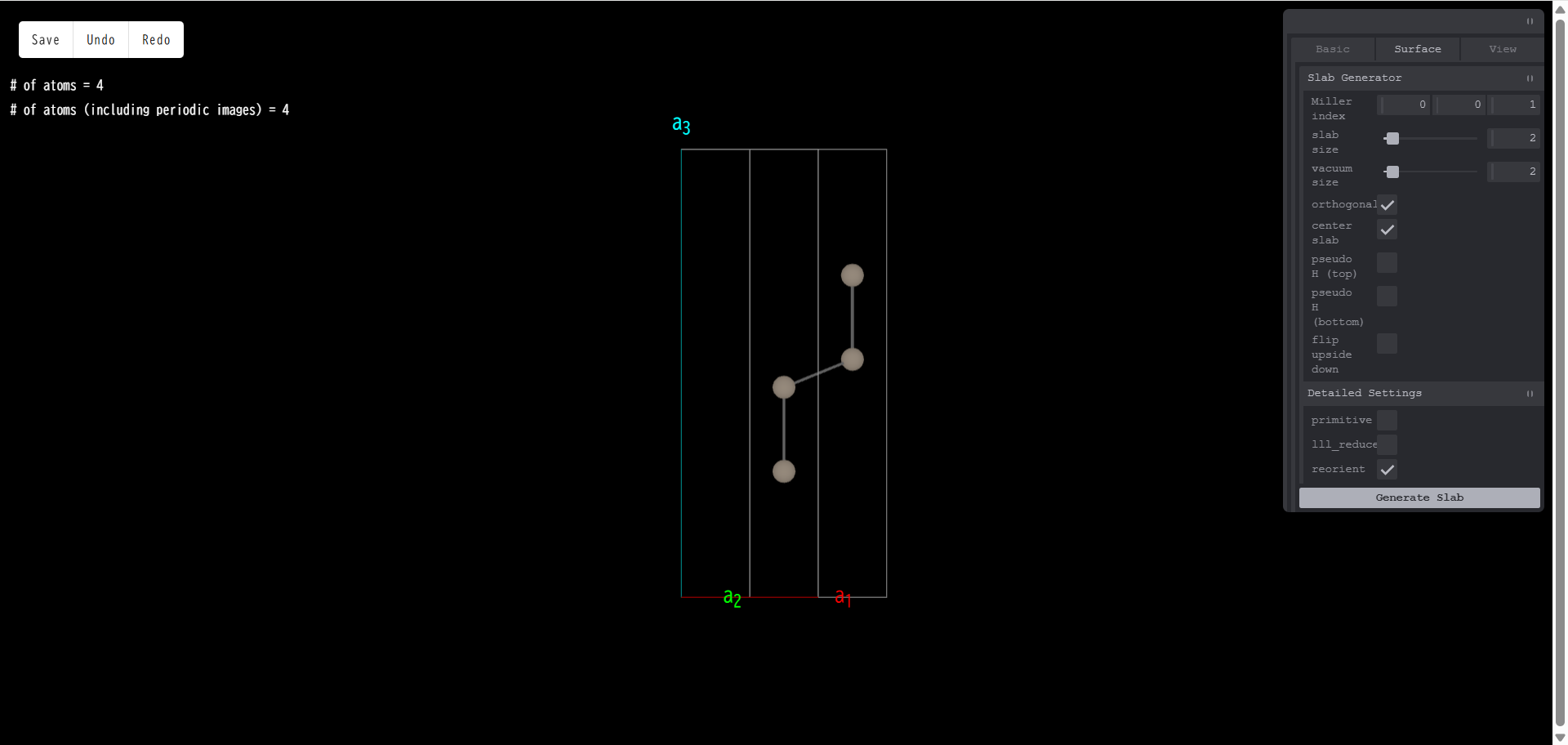
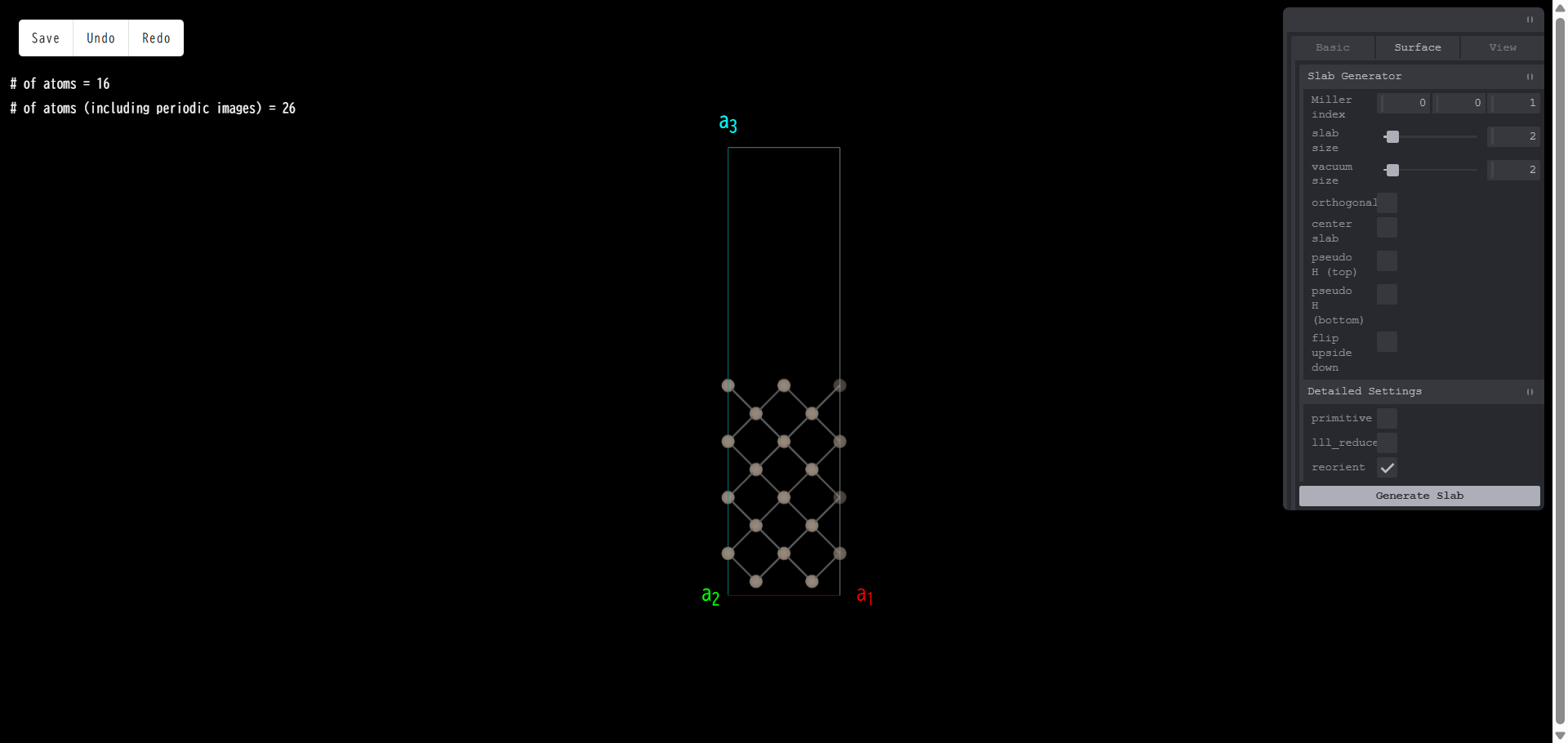
center slab:スラブ領域をセルの中央に配置します。デフォルトでチェックが入っているので、チェックを外し、「Generate Slab」を押すと、次のようになります。
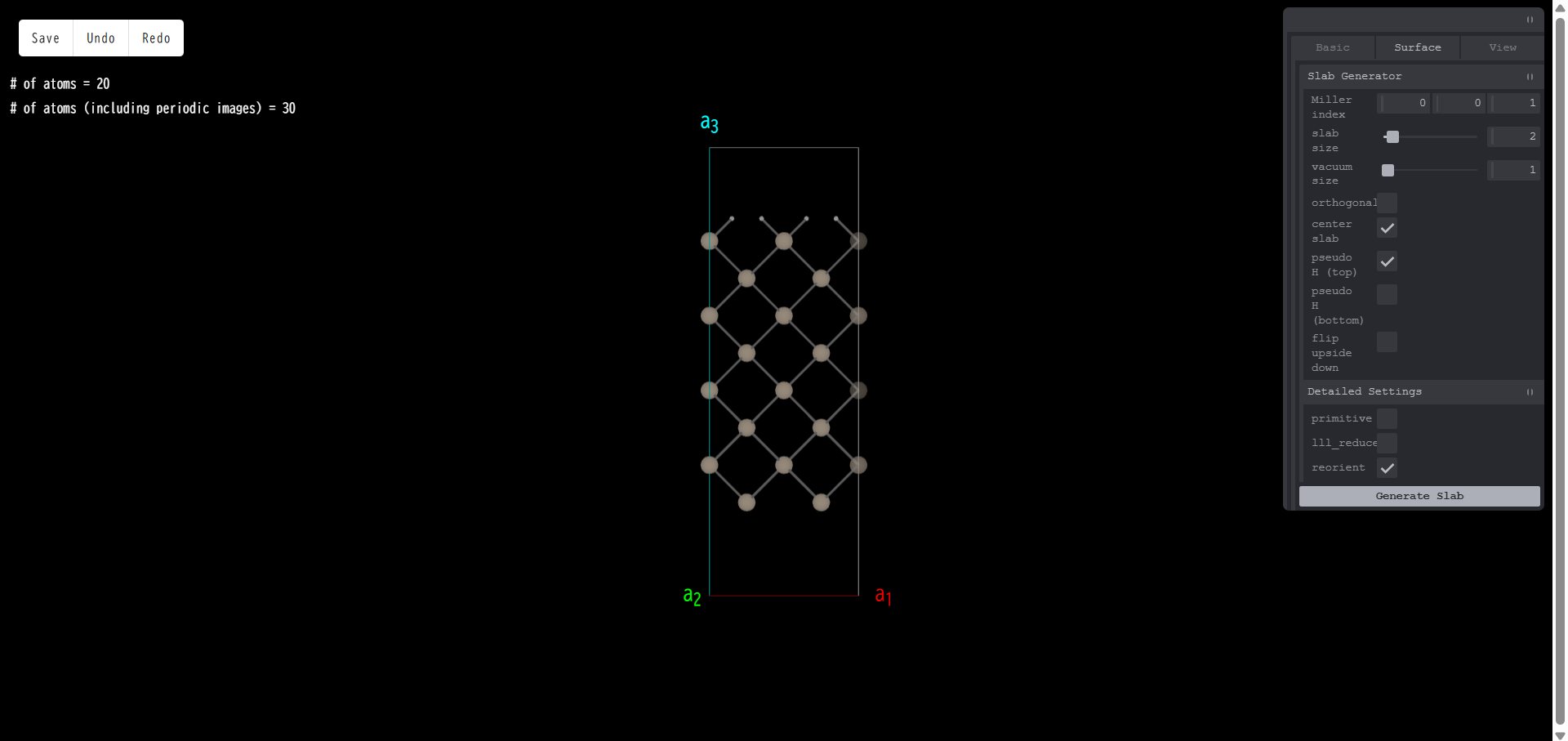
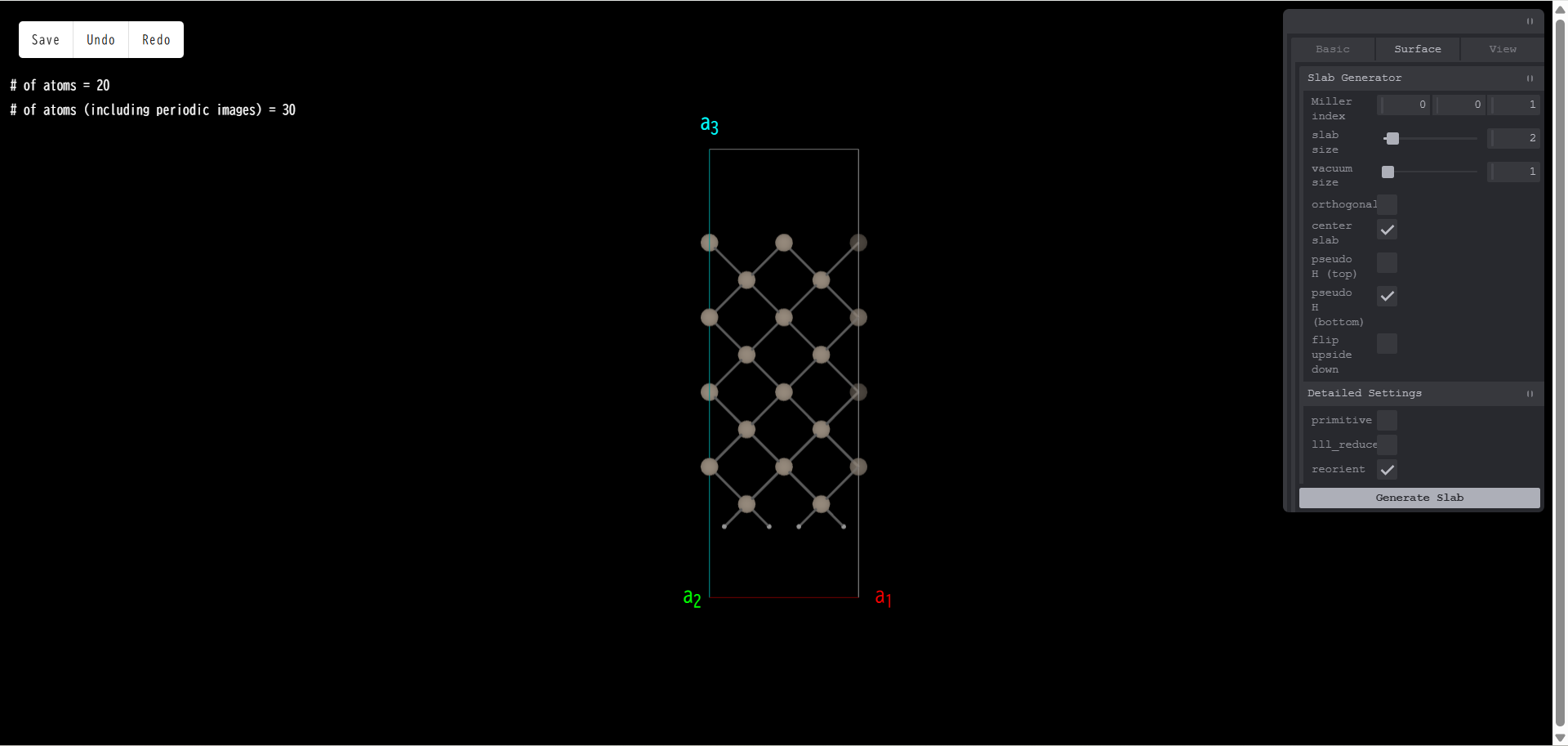
pseudo H:終端用の擬水素原子を、表面の上部または下部に追加します。Z=1 以外の電荷を設定したり、表面原子との結合距離を設定することが可能です。
flip upside down:スラブモデルの上下を反転させます。例として、以下のようなモデルを作成します。
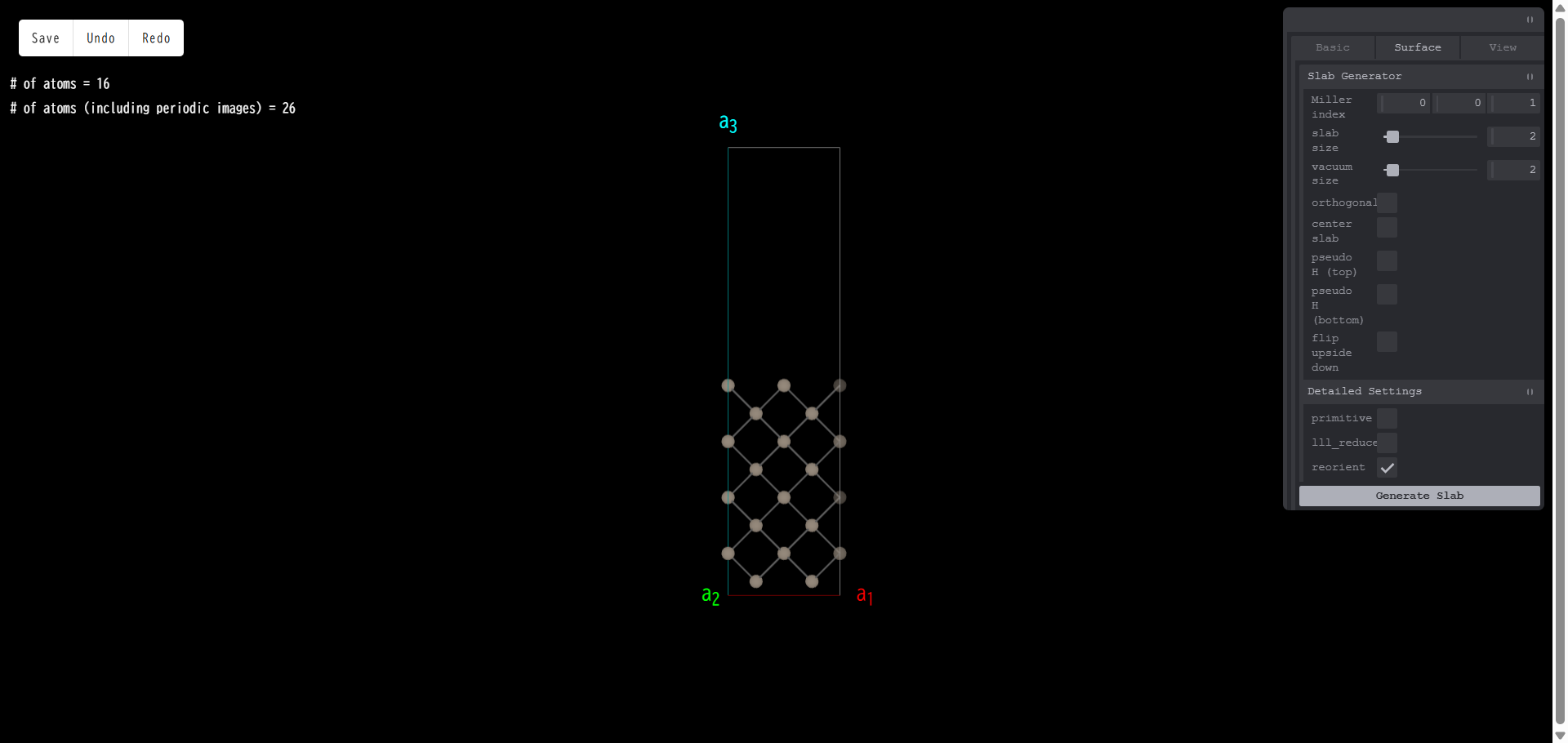
ここで「flip upside down」にチェックを入れ、「Generate Slab」を押すと、次のようになります。
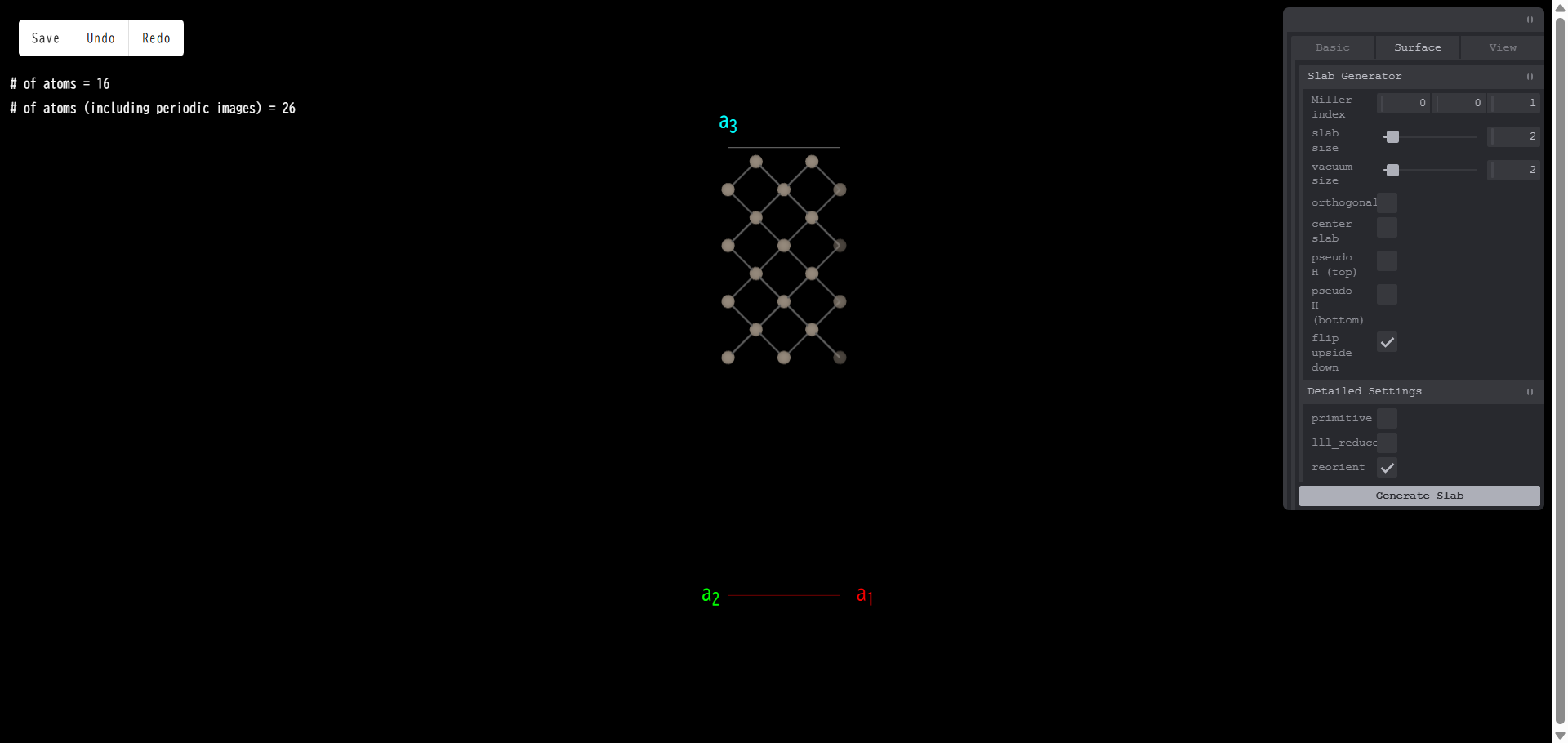
primitive:スラブモデルを primitive セルに変換します。例として、以下のようなモデルを作成します。
上面図は以下の通りです。
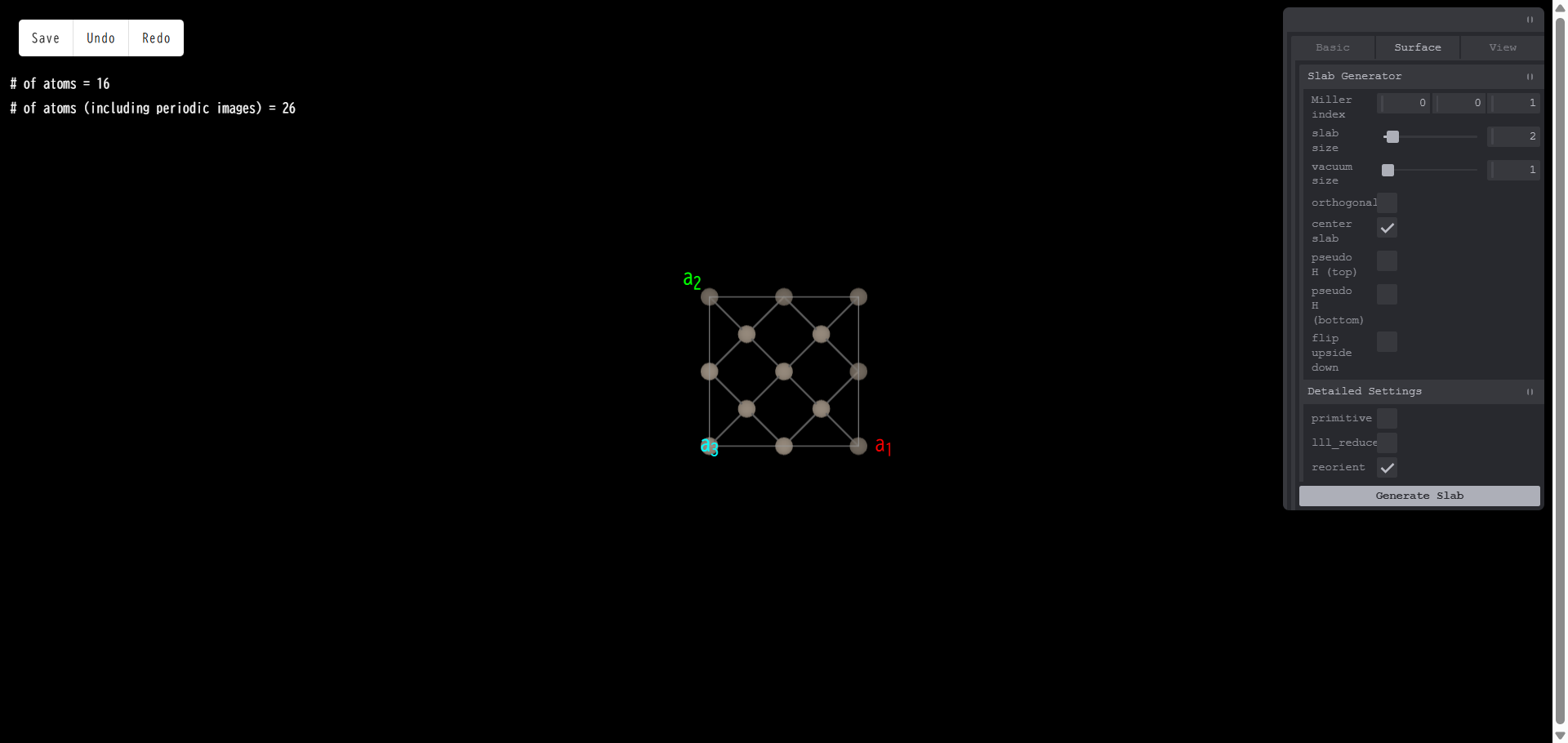
ここで「primitive」にチェックを入れ、「Generate Slab」を押すと、側面図は次のようになります。
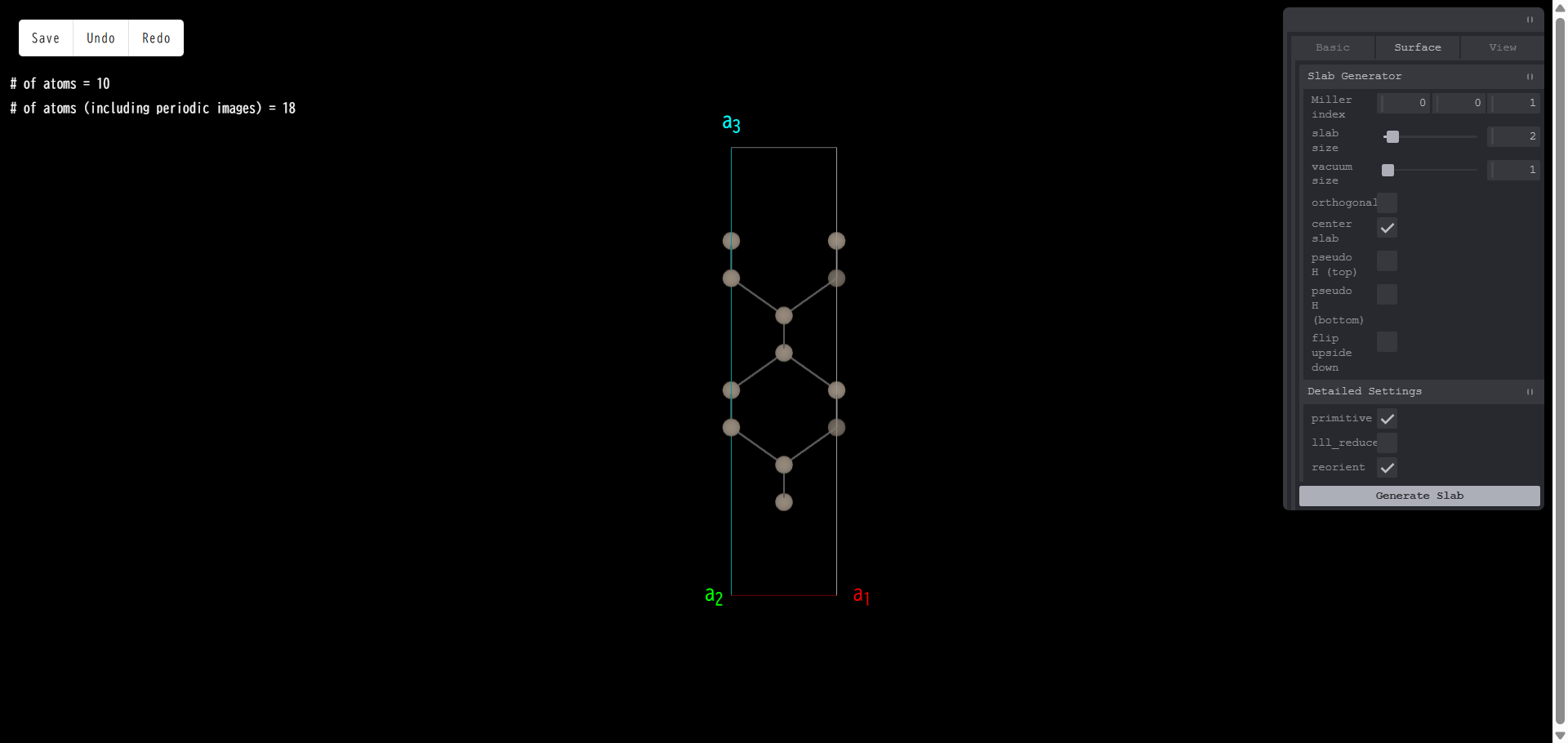
上面図を見ると、モデルが primitive セルに変換されていることが分かります。
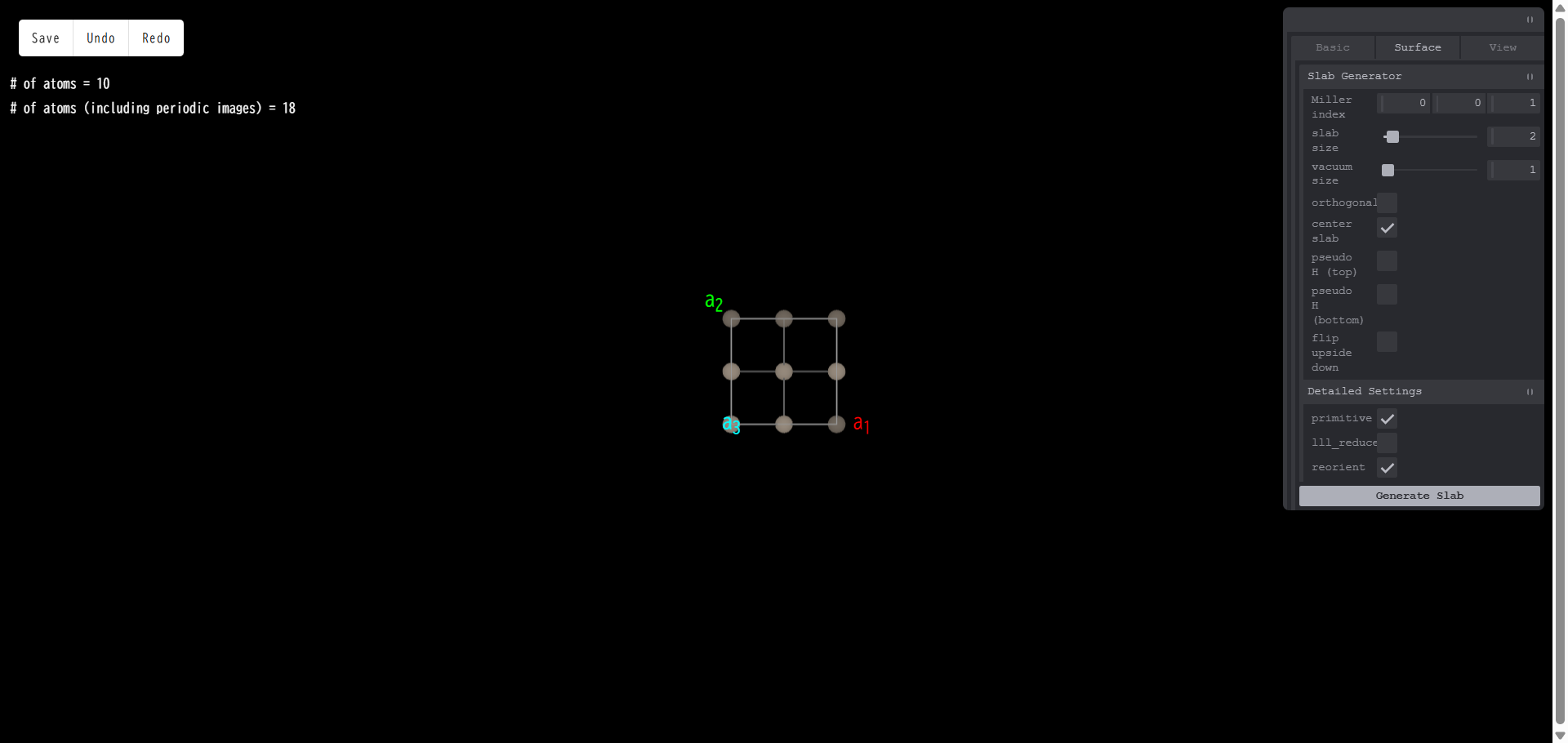
lll_reduce:「orthogonal c slab」とは異なり、原子構造は変化させずに、ベクトル a3 を表面になるべく垂直にとる機能です。例として、以下のようなモデルを作成します。
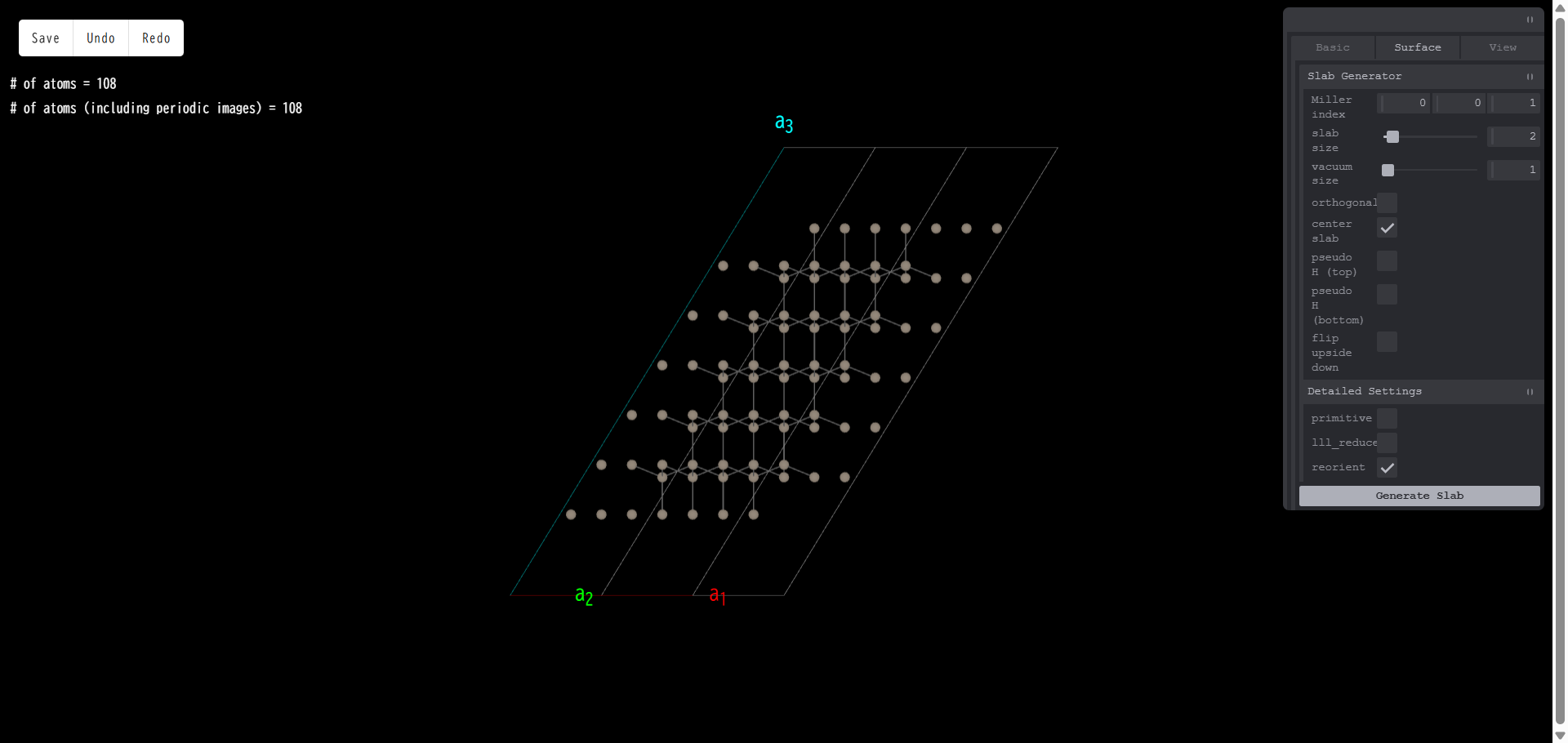
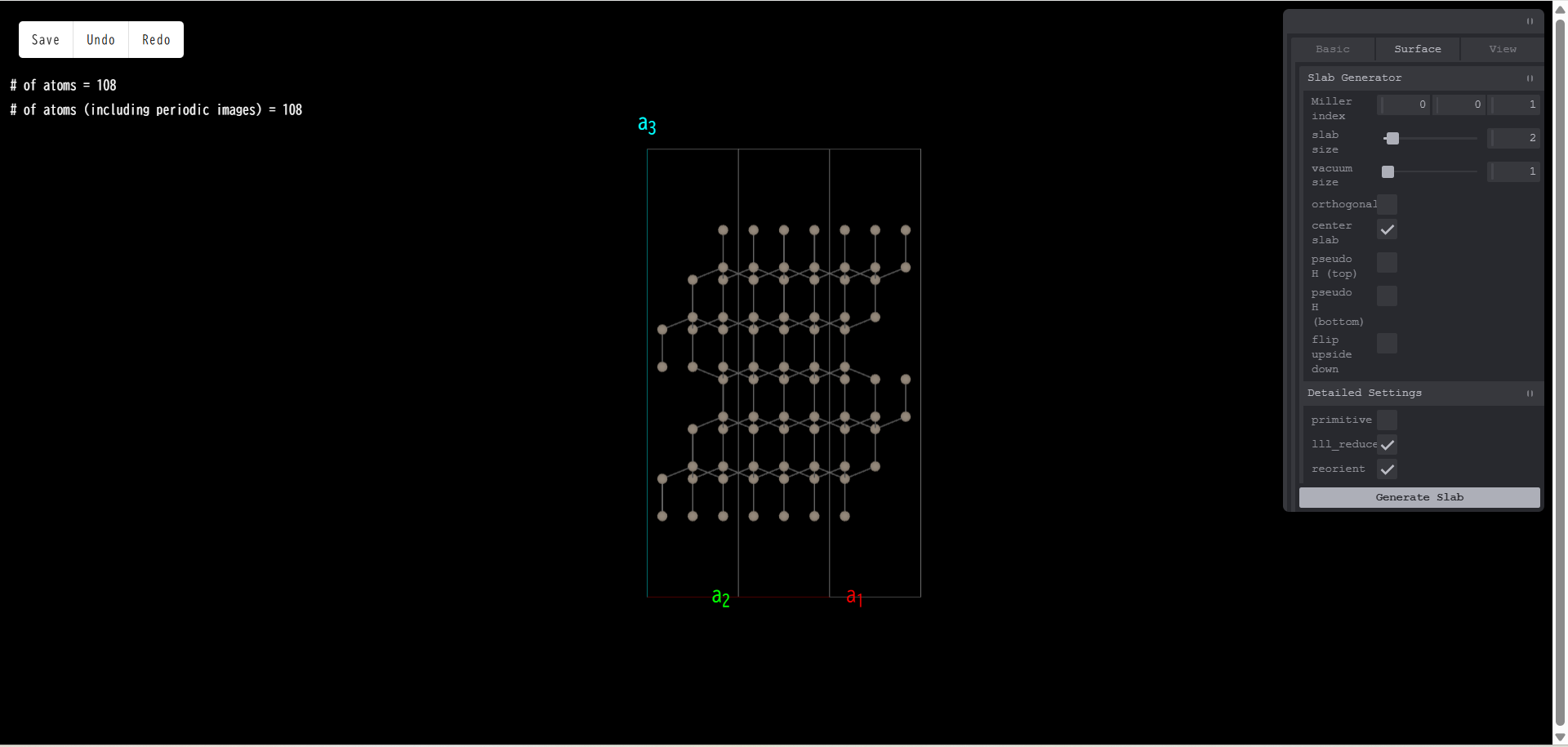
ここで「lll_reduce」にチェックを入れ、「Generate Slab」を押すと、ベクトル a3 が垂直に近づいています。
これらのパラメータの詳細については pymatgen(https://pymatgen.org)の SlabGenerator に関するドキュメントをご参照ください。
7.10. フッターメニュー
フッターメニューにある「Add Crystal」ボタンをクリックすると、Quloud に登録されている(同じ Project 内にある) 別の Material(結晶)の一覧が表示されます。
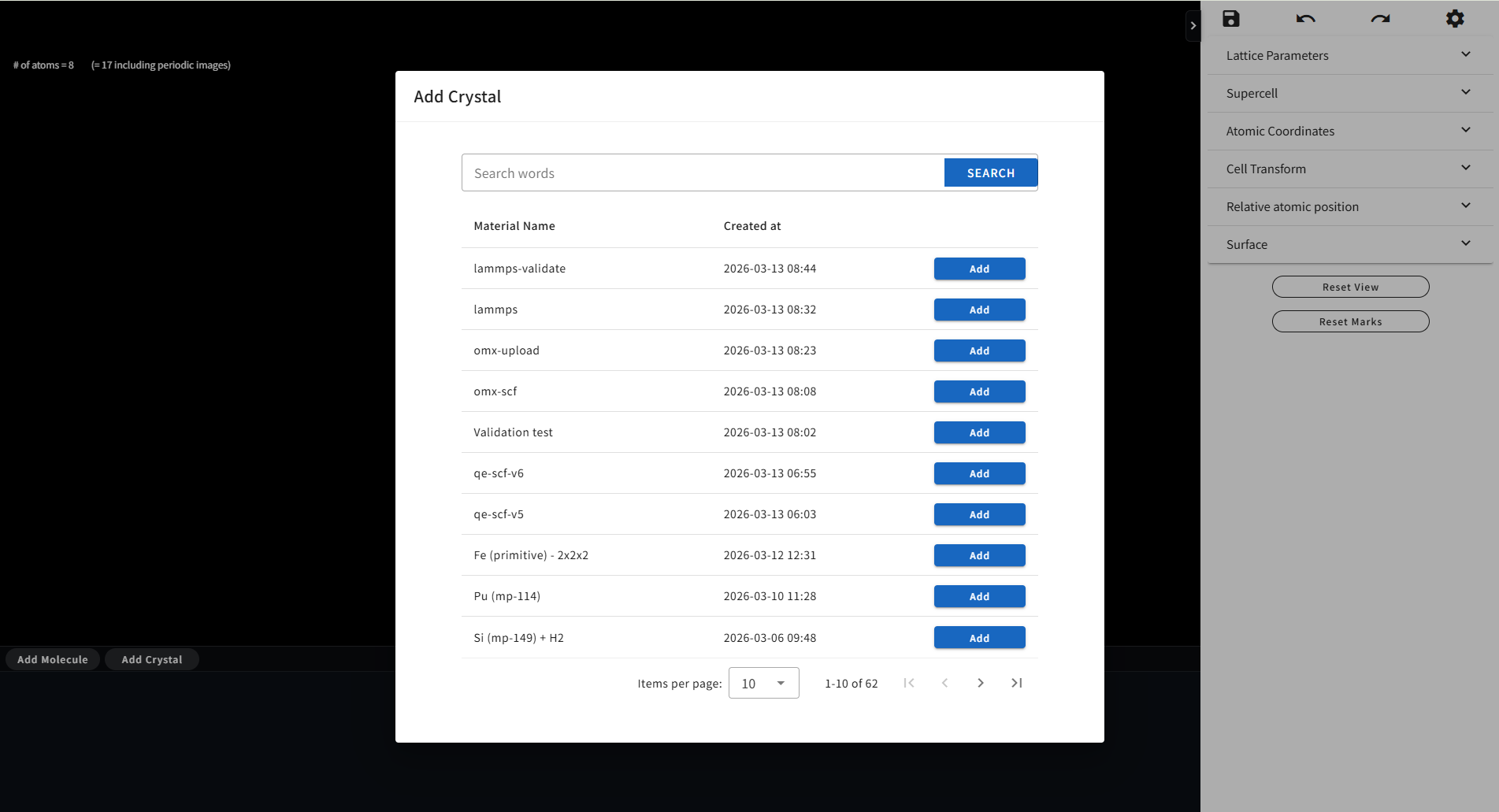
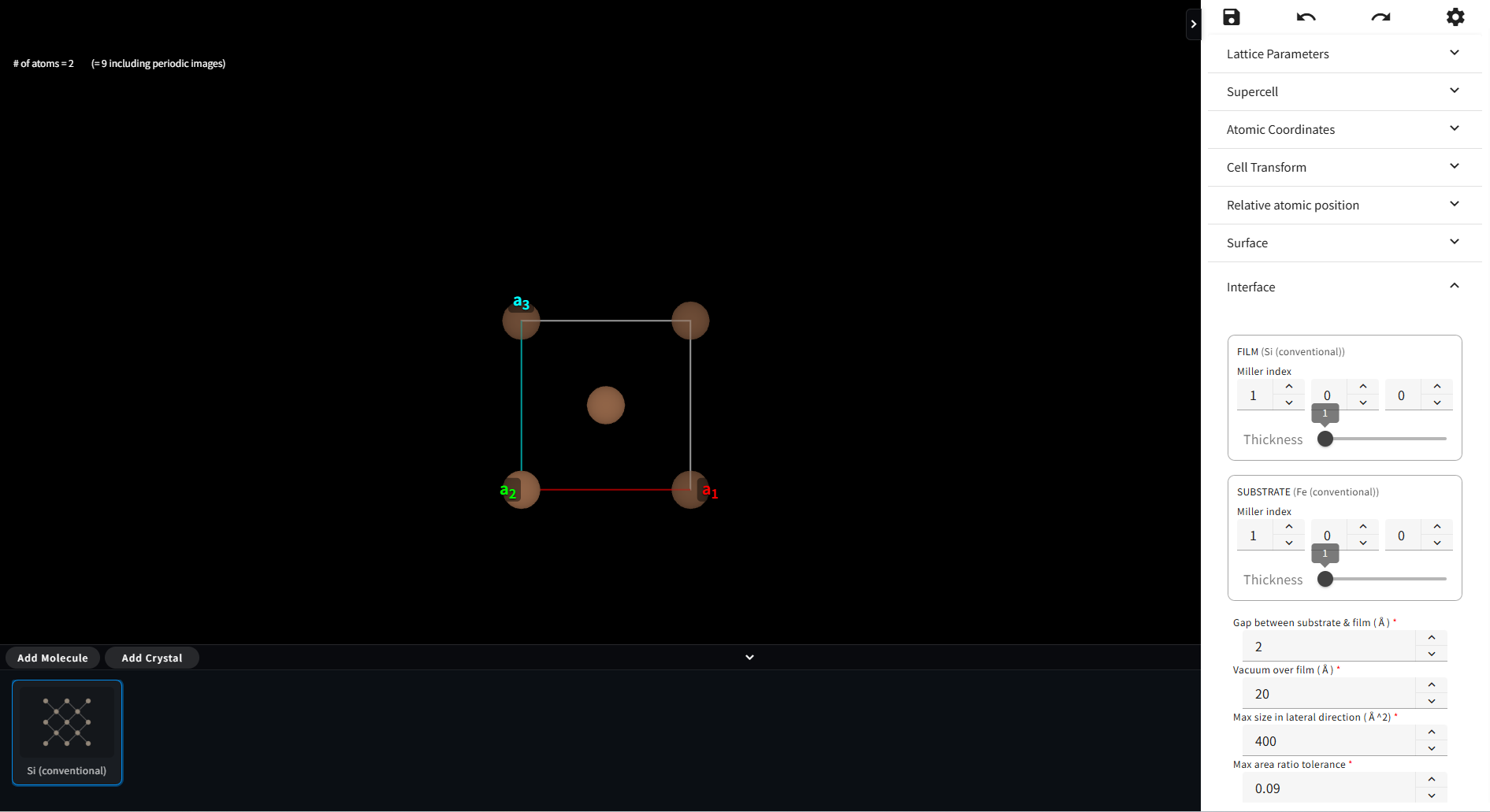
この中から、現在モデリングを行っている Material と組み合わせて界面モデルを作るためのペアとなる相手を選びます。 候補が多数ある場合は、上部の Serach を使って検索することもできます。 相手が見つかったら「Add」ボタンをクリックします。すると、下図のように、フッターメニュー内にその結晶が登録されます。 同様にして複数の結晶を登録することも可能ですが、モデリングを行うペアとして選択できるのは一つだけです。
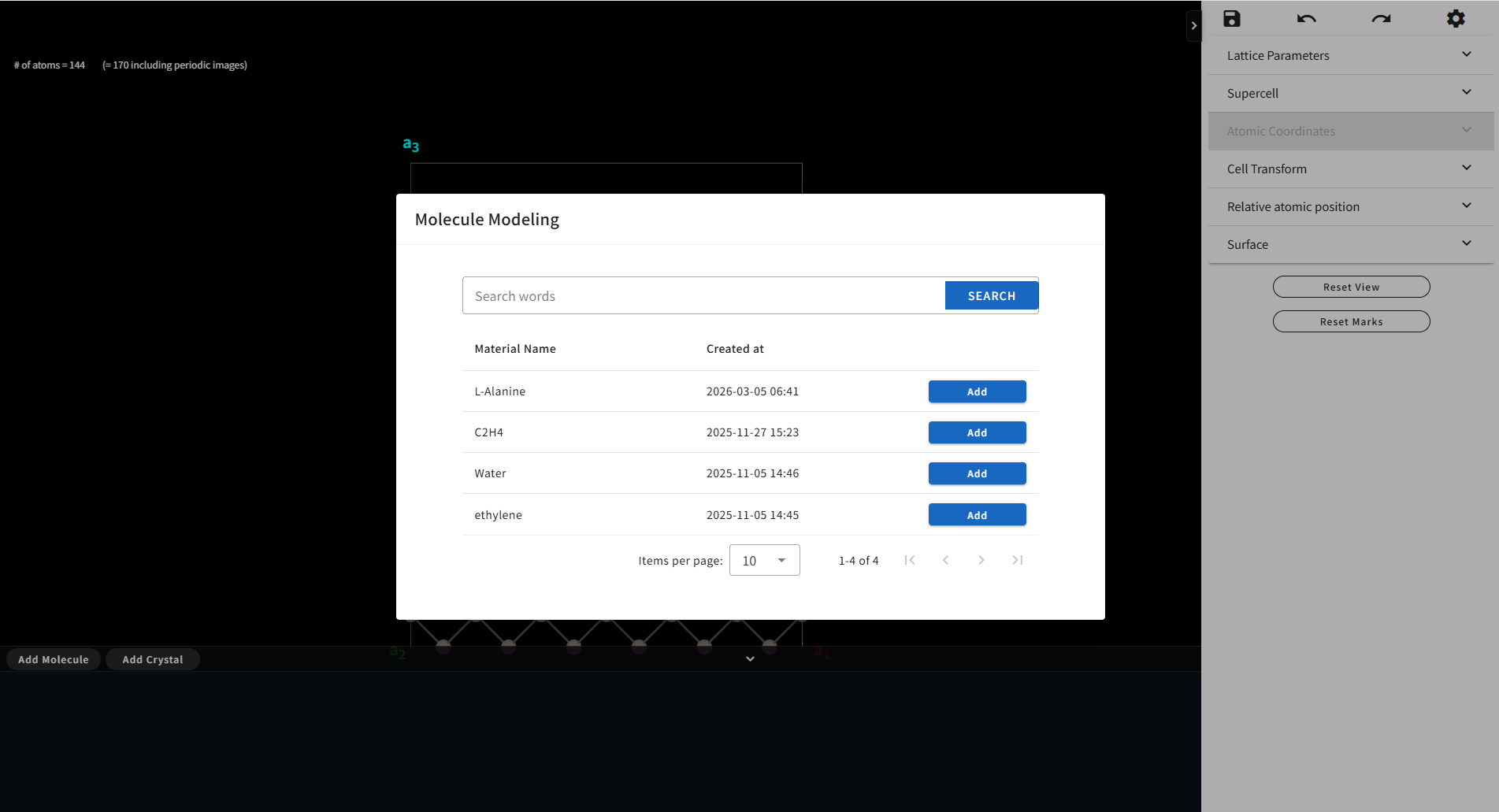
フッターメニューにある「Add Molecule」ボタンをクリックすると、Quloud に登録されている(同じ Project 内にある) Material(分子)の一覧が表示されます。
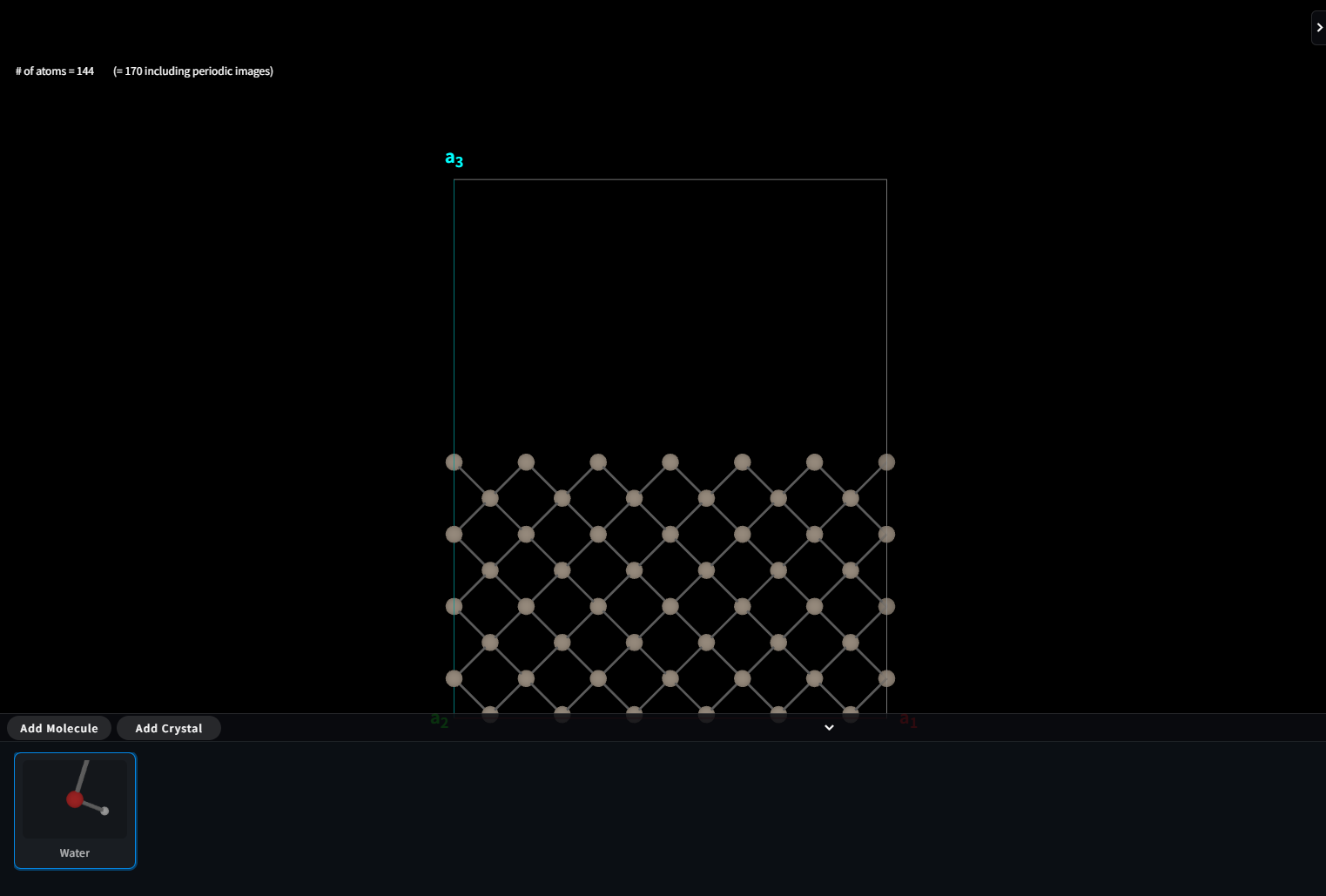
この中から、現在モデリングを行っている Material(結晶) と組み合わせて、結晶中への分子の挿入や、 表面上の分子吸着モデル、スラブモデルの真空領域の一部を分子集団で充填する、固液界面や固気界面のモデル作ることができます。 相手が見つかったら「Add」ボタンをクリックします。すると、下図のように、フッターメニュー内にその分子が登録されます。 同様にして複数の分子を登録することも可能ですが、モデリングを行うペアとして選択できるのは一種類だけです。
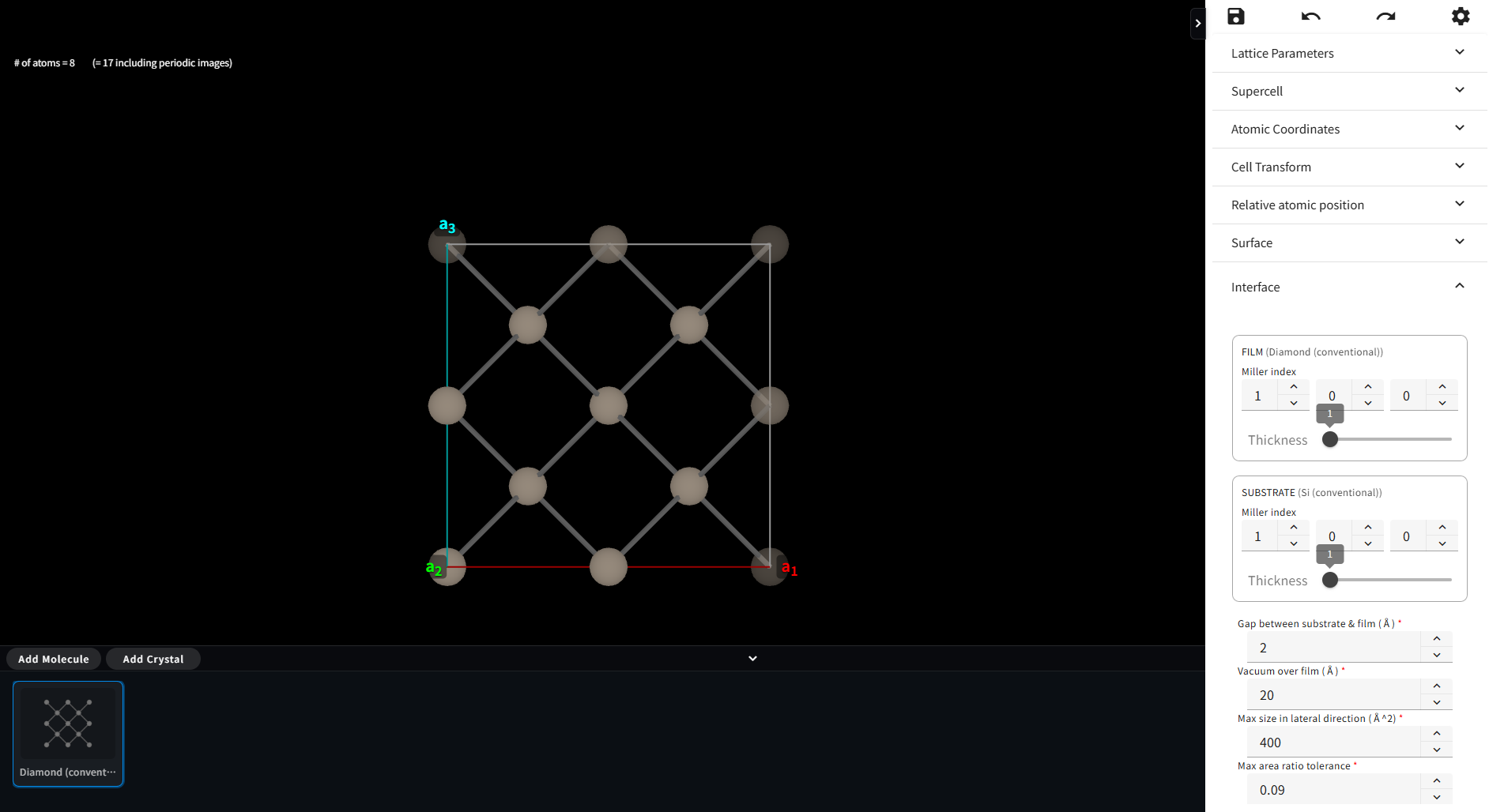
7.11. Interface
「Add Crystal」でフッターに登録された結晶の一つが選択状態になると、右サイドパネルに Interface というメニューが現れます。
Interface メニュ-では、現在モデリング中の結晶が基板(Substrate)、フッターで選択された結晶をフィルム(Film)として、 それぞれの Material の結晶面方位やスラブの厚さ等が設定できます。 フィルム側と基板側の格子定数をなるべく歪なくマッチさせるように界面を作るため、 探索範囲を面積で制限したり、歪の許容範囲等を指定する必要もあります。設定メニューでは
フィルムのミラー指数、および膜厚の指定(Film Miller & Thickness)
基板のミラー指数、および膜厚の指定(Substrate Miller & Thickness)
フィルム、基板間の初期ギャップ(Gap between substrate & film)
真空領域のサイズ(Vacuun over film)
界面構造の候補を探索する面積の範囲(Max size in lateral direction)
界面構造の候補を探索する際に、フィルムと基板の面積のずれをどこまで許容するかの範囲(Max area ratio tolerance)
界面構造の候補を探索する際に、フィルムと基板の格子ベクトル長さのずれをどこまで許容するかの範囲(Max length tolerance)
界面構造の候補を探索する際に、フィルムと基板の格子ベクトル間角度のずれをどこまで許容するかの範囲(Max angle tolerance)
といった設定が可能です。設定後「Submit」ボタンを押すと界面構造の最初の候補と、 候補の切り替え、および界面モデルを確定するための小パネルが表示されます。
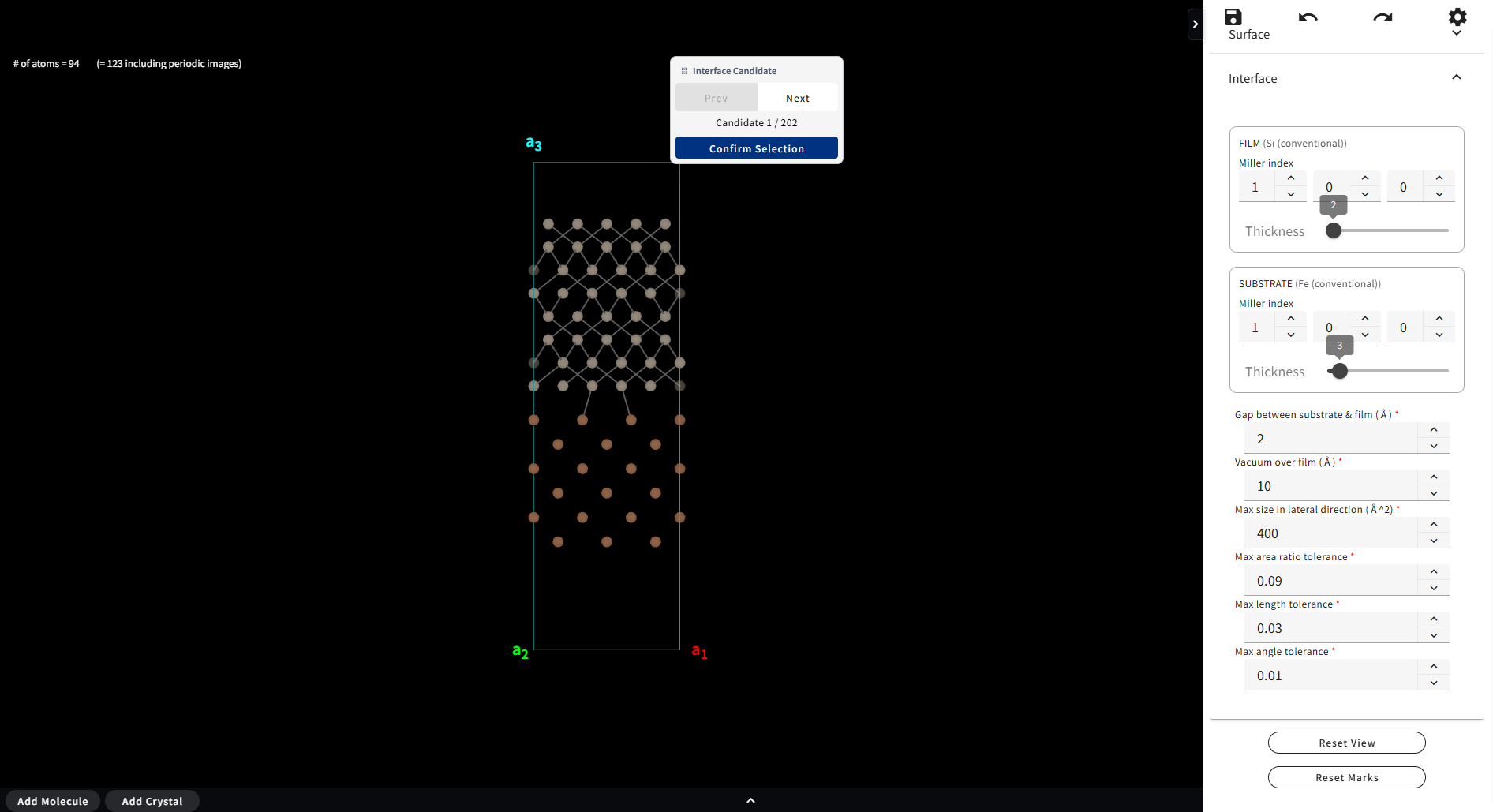
最初の方の候補がより原子数の少ないモデルになっています。 「Confirm Selection」で界面モデルを確定させたら、その後は通常のモデリング画面に戻ります。
7.12. Add Molecule
フッターの「Add Molecule」ボタンをクリックして登録された分子の一つが選択状態になると、 右サイドパネルに Add Molecule および Packmol というメニューが現れます。 ここでは、バルクやスラブといった周期構造に孤立分子を挿入したモデルを作成するための Add Molecule について説明します。

Add Molecule メニュー内の「Add Molecule」ボタンをクリックすると、下図のような画面になります。
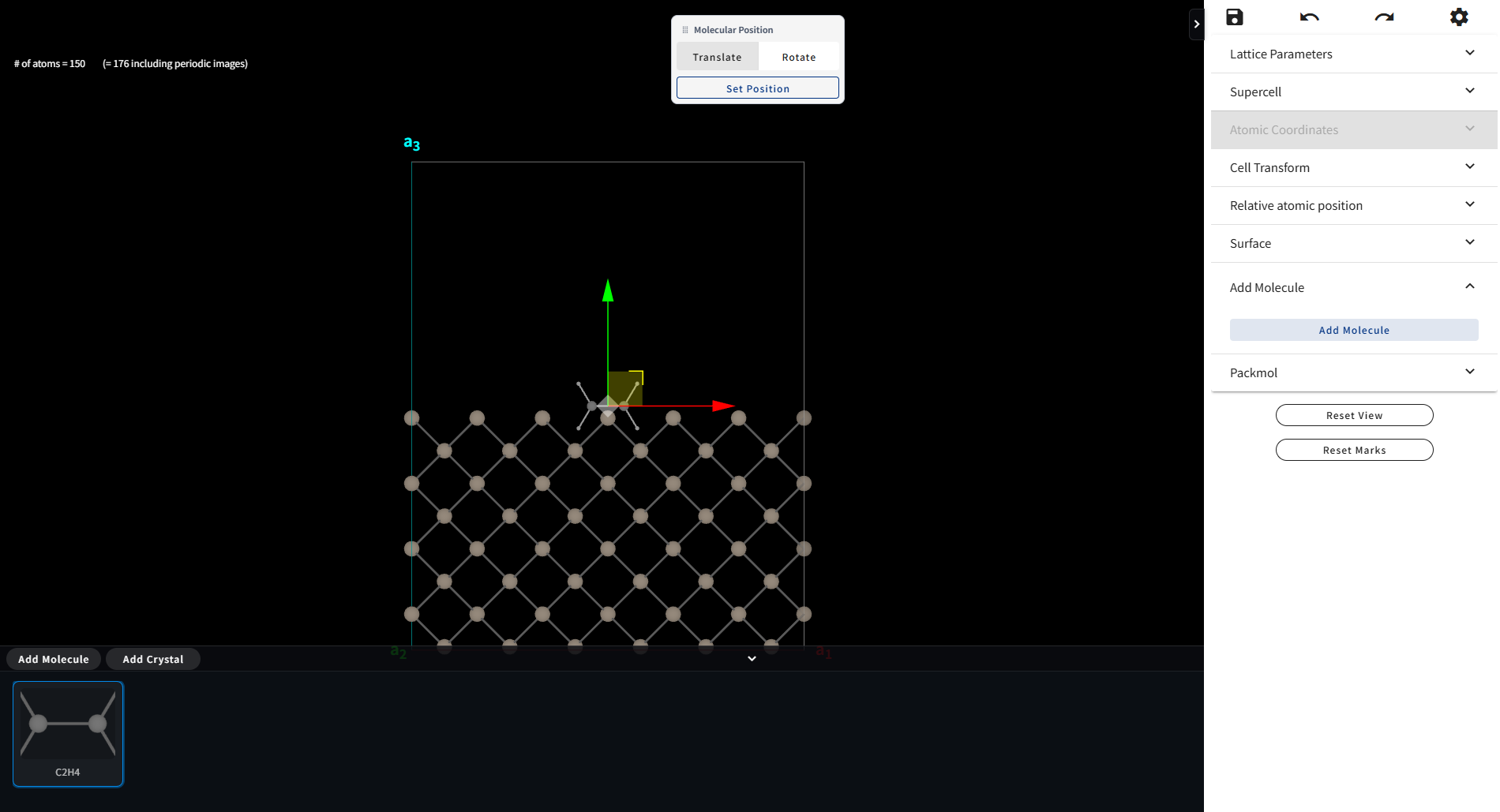
追加した分子には赤、青、緑の3本の矢印が表示されており、 いずれかの矢印をドラッグすると、その矢印の方向に分子を平行移動させることができます。 また小さな四角の部分をドラッグすると、2次元的平行移動させることもできます。
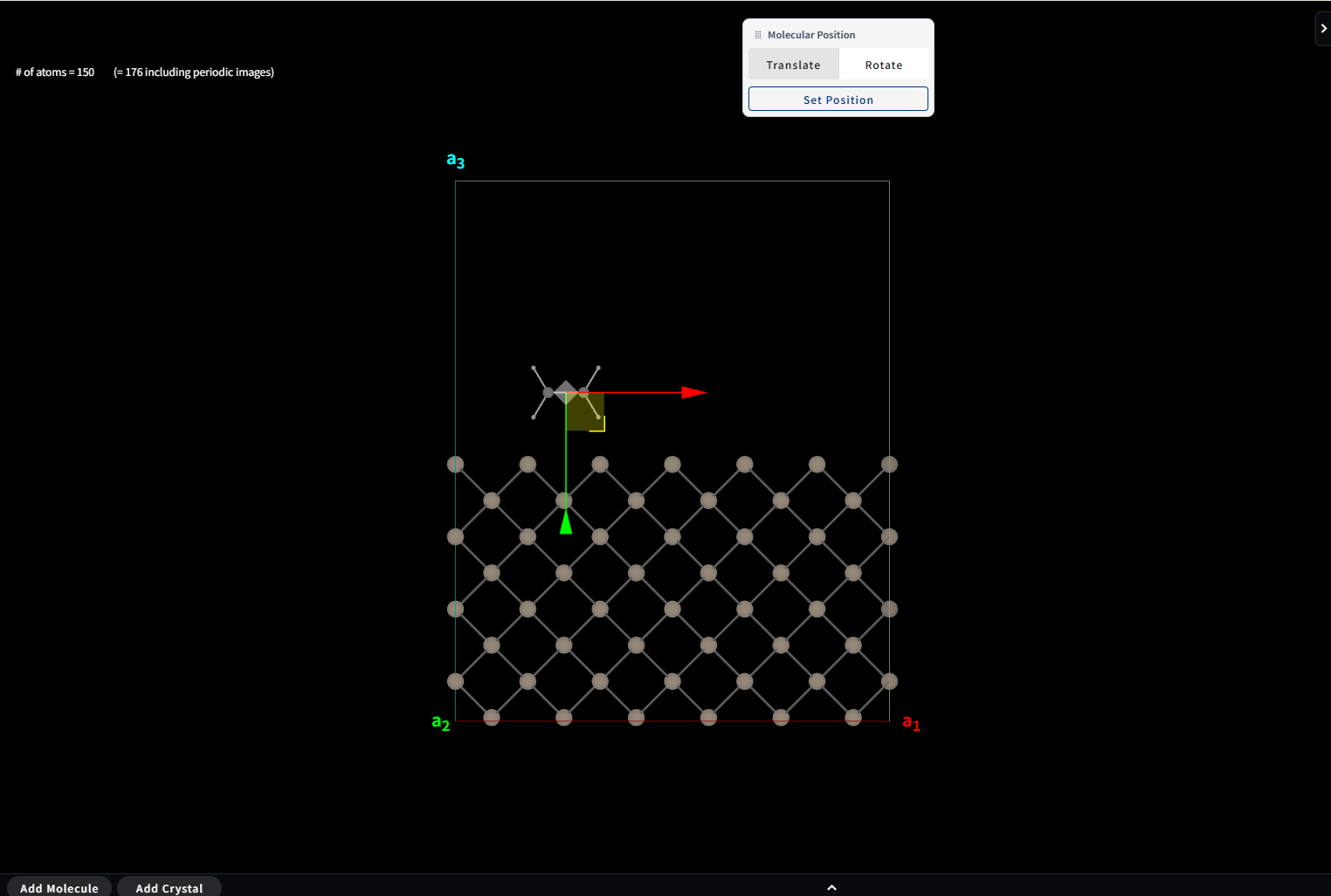
Add Molecule を押した直後は Translate モードですが、画面上部の小パネルで、Rotate モードに切り替えることができ、 今度は分子を回転させることができるようになります。
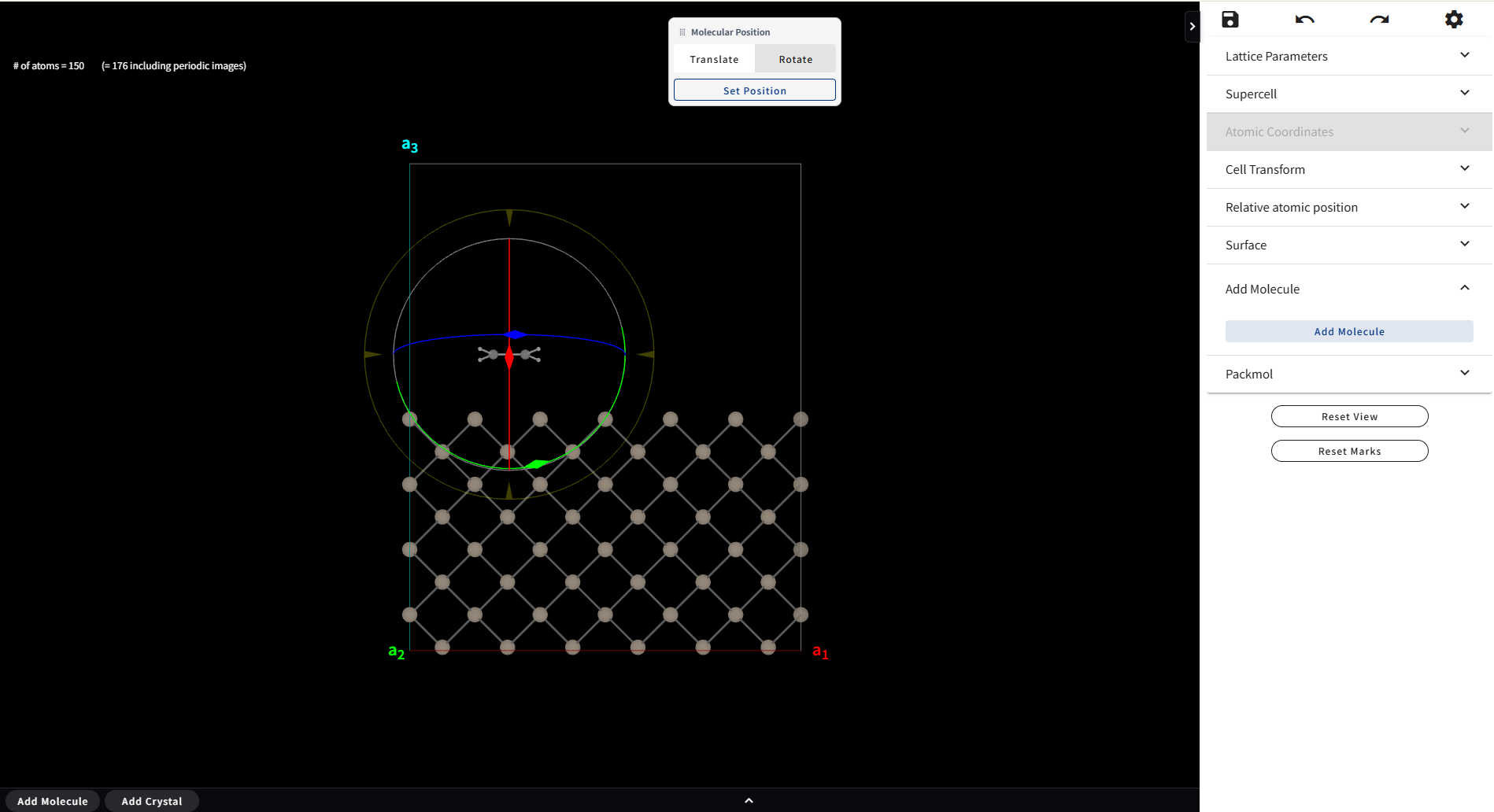
位置と回転が決まったら、最後に小パネル内の「Set Position」をクリックします。 そうすると通常のモデリング画面に戻ります。
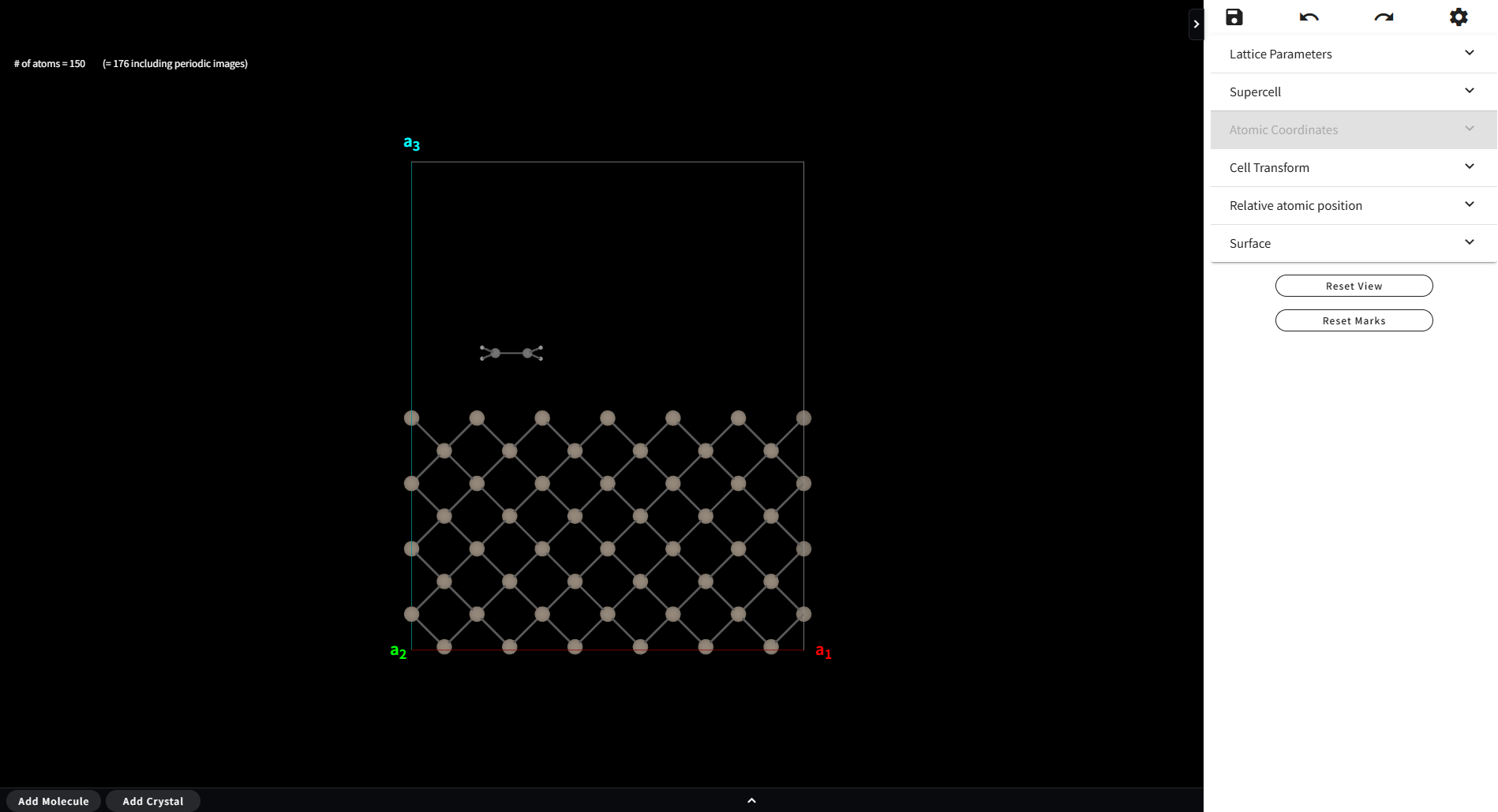
7.13. Packmol
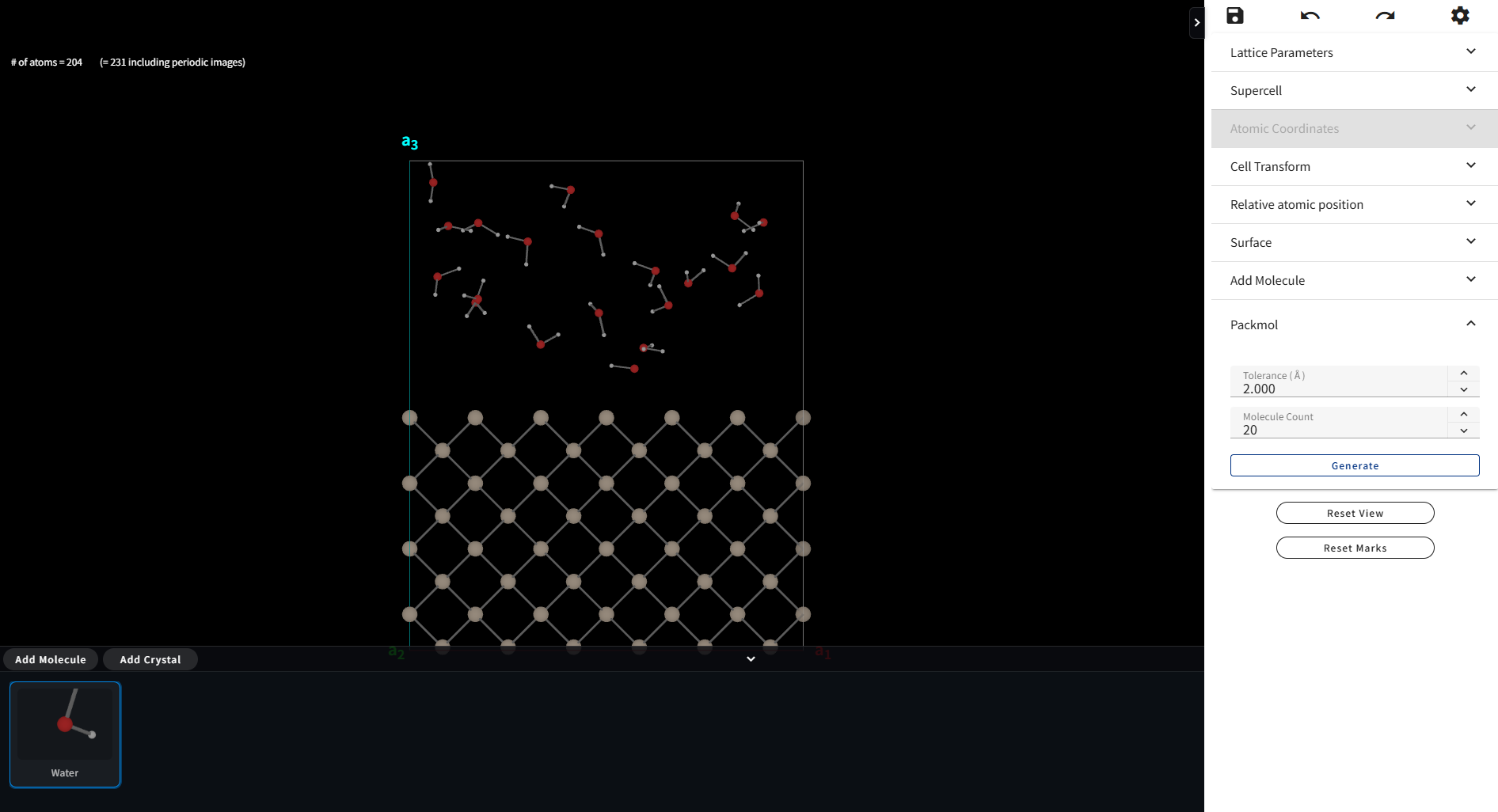
ここでは、フッターの「Add Molecule」ボタンをクリックして登録された分子の一つが選択状態になったときに現れる Packmol というメニューについて説明します。Packmol は、https://m3g.github.io/packmol/ で保守されているソフトウェアで、 指定した密度で分子をある領域に詰め込む(Packing)モデリングを行うものです。 Quloudでは、下図のように真空領域を持つモデルの、真空領域部分に分子を詰め込むのに利用します。

Packmol メニューでは、
分子および結晶間の距離ある一定以上に保つパラメータ(Tolerance (Å))
詰め込む分子の個数(Molecule Count)
が設定できます。設定後に「Generate」ボタンをクリックすると、下図のようなモデルが作れます。
警告
真空領域のサイズに比べて分子の個数が多すぎると、一定距離をあける条件を満たせなくなり、モデリングが上手くいかないことがあります。
ちなみに作られたモデルは周期境界条件のため、実際には下図のような状況になっていることに気を付ける必要があります。
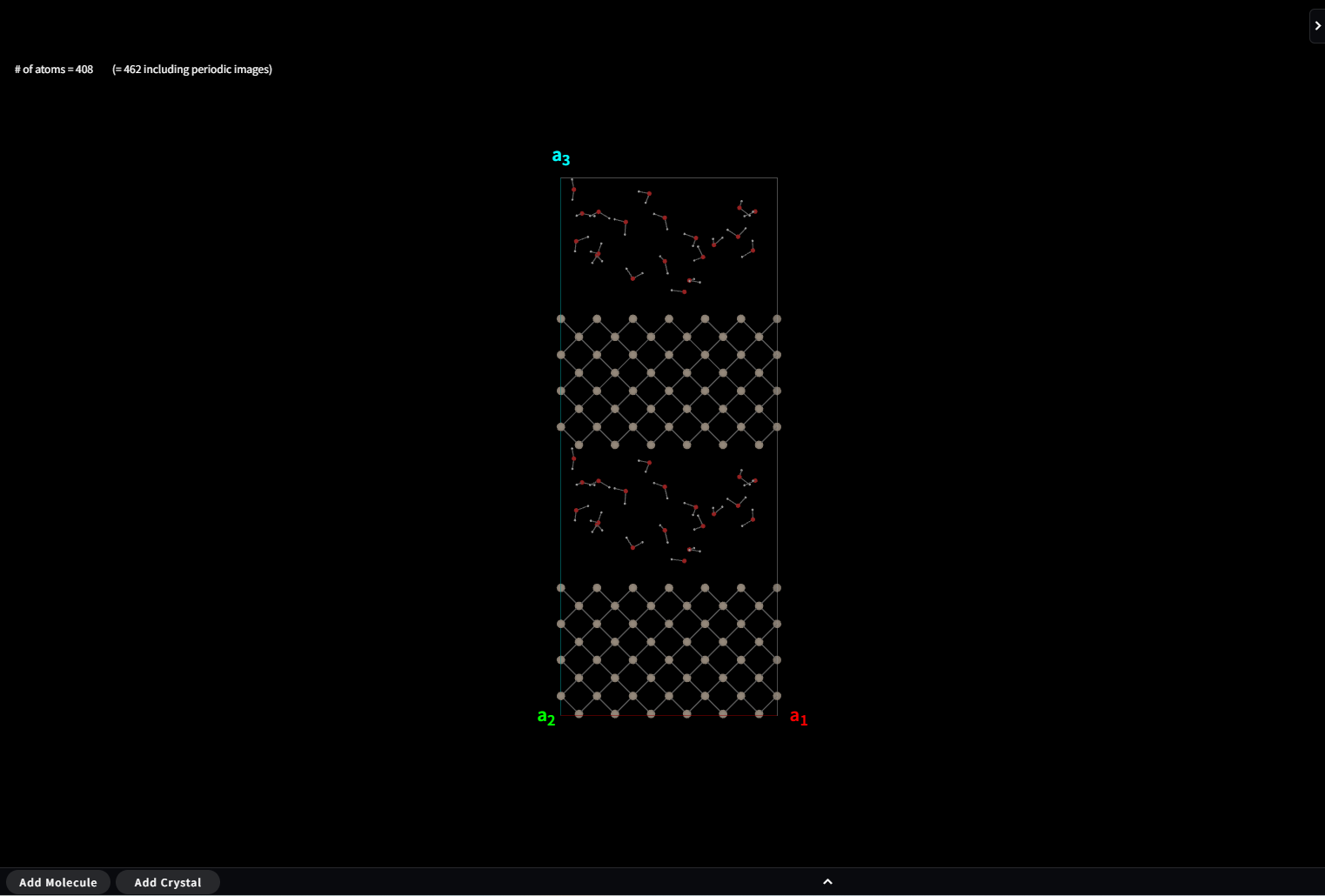
7.14. Add Cell
これは、モデリングを開始した Material が Molecule の場合にだけ現れるメニューです。 通常、孤立分子の原子座標データ(例えば xyz 形式)は、ユニットセルの情報を持たないため、 これにユニットセルの情報を追加してスーパーセルモデルを作成し、周期境界条件が課されるタイプの計算ソフトで扱えるようにするための機能です。
Add Cell メニュー内にある「Add Cell」ボタンをクリックすると、下図のようになります。
上図の右側操作パネルの内容からもわかるように、Add Cellで作られた分子のスーパーセルモデルは、 以降、結晶(周期系)として扱われることになります。
7.15. パネル上部アイコン
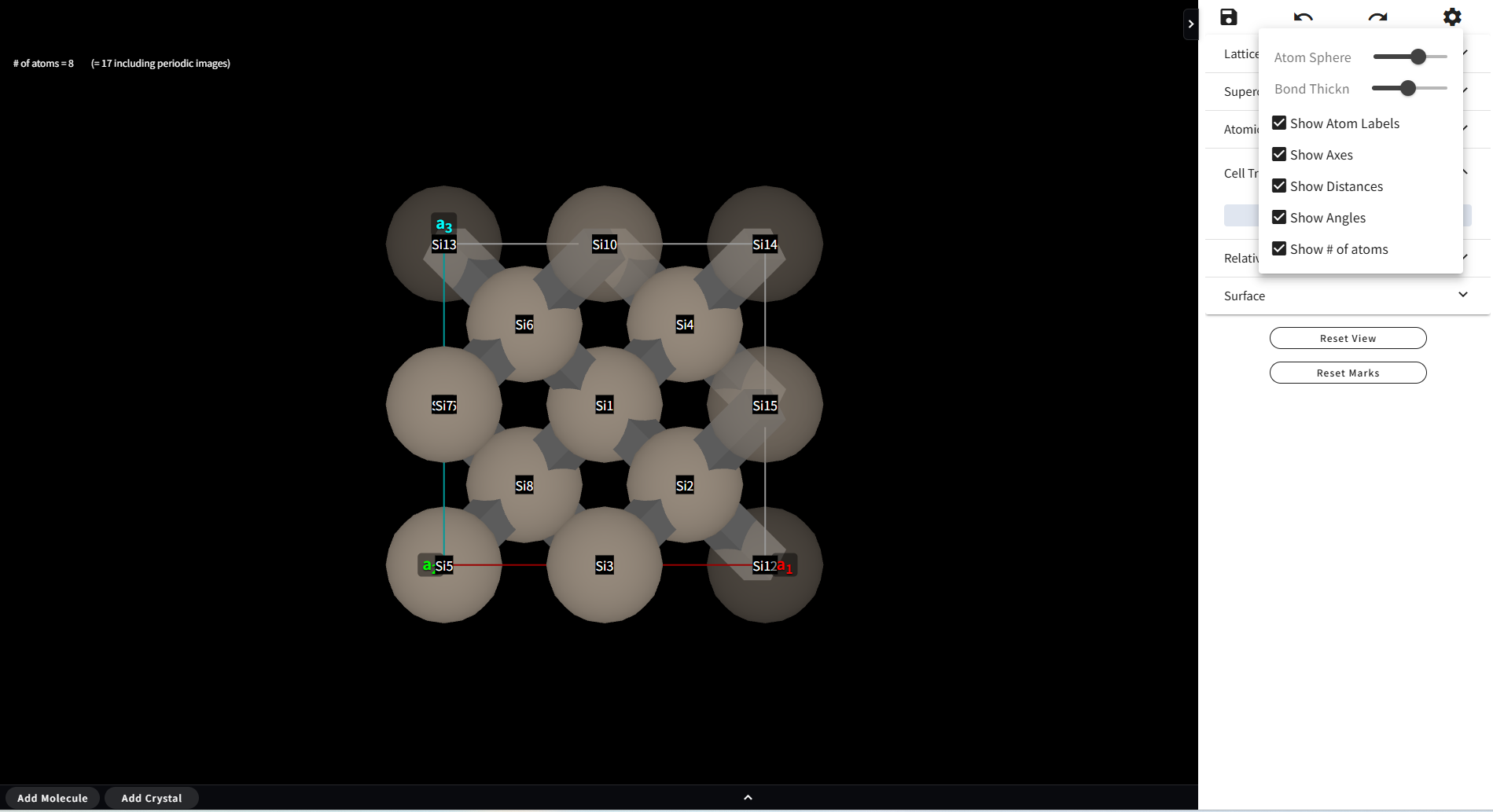
モデリング画面右側の操作パネル上部に「Save」「Undo」「Redo」「Settings」の4つのアイコンがあります。 このうち、Undo / Redo については、モデリングの操作を一つ前に戻す、あるいは進める、 というもので、改めてご説明する必要もないと思われます。 Settings は、原子のサイズや、元素名のラベルを表示させるなど、表示に関する設定をまとめたもので、 例えば下図のような見た目に変えることができます。
7.16. モデルの保存
操作パネル上部の4つのアイコンのうち、一番左にあるのが「Save」で、こちらから 作成したモデルを Material として保存(Quloudに登録)する事ができます。 クリックすると、次のようなメニューが開きます。
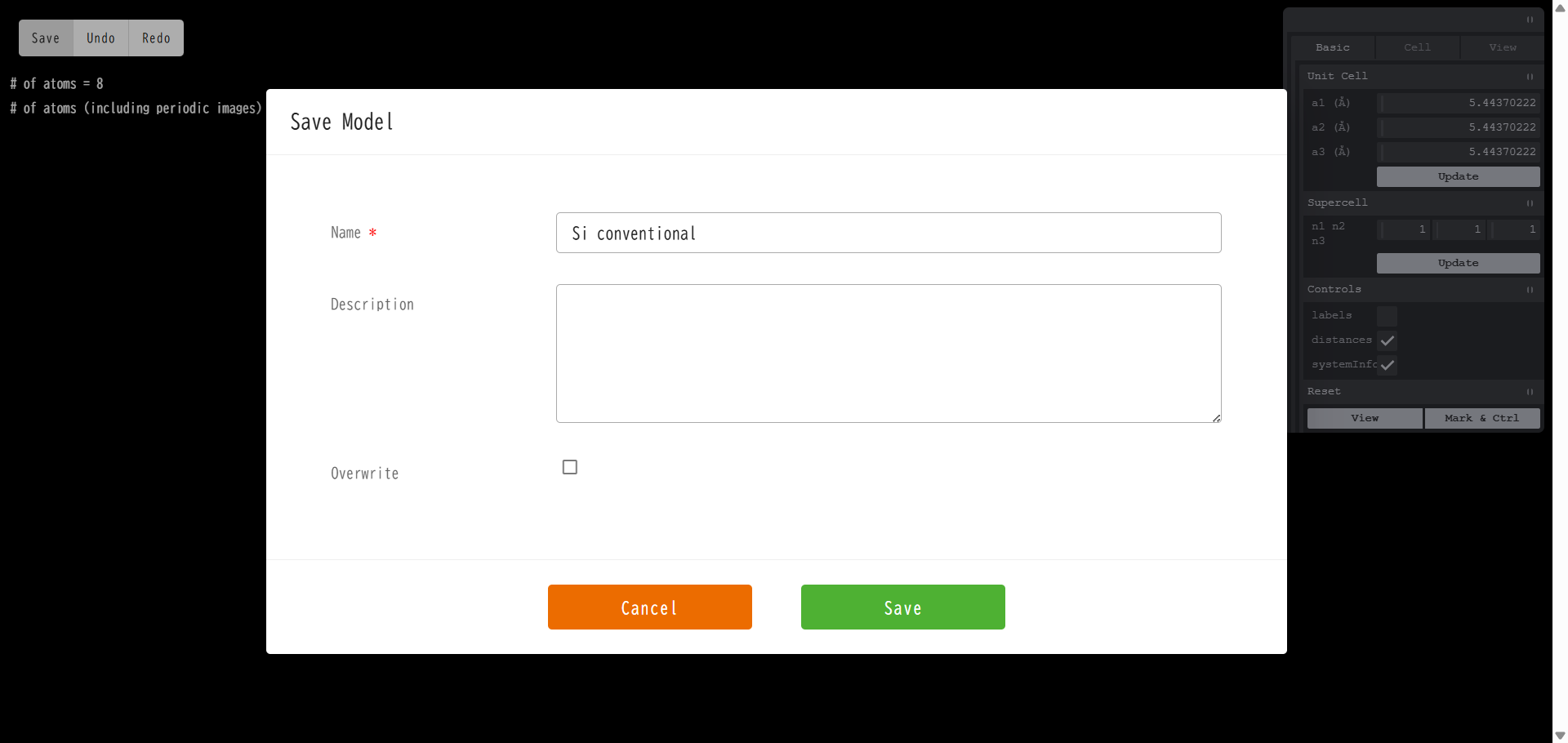
保存するモデルの名前は必須入力となっています。デフォルトでは、モデルは新規の Material として保存されますが、 「Overwrite」にチェックを入れると、モデリングを開始したときの Material に上書き保存されます。
警告
すでにその Material で Job 作成を行っている場合、または Interface, Add Molecule, Add Cell のモデリングを行った場合には、 Overwrite はできないようになっています。
最後に「Save」をクリックします。確認ダイアログが出るので「OK」とすると保存が完了し、新しい Material 詳細画面に移ります。